发病机制:
本病的发病机制目前尚不清楚,有非单一遗传背景,国外文献报道家族发病率为44%,国内有报道为11%。有研究发现,儿童发病与Xq28染色体G4.5基因突变有关,成人发病与常染色体11p15关系密切。此外,肿瘤坏死因子转换酶异常、心内膜下心肌缺氧以及多种致畸因素均可能参与本病的发生。
一、病理解剖
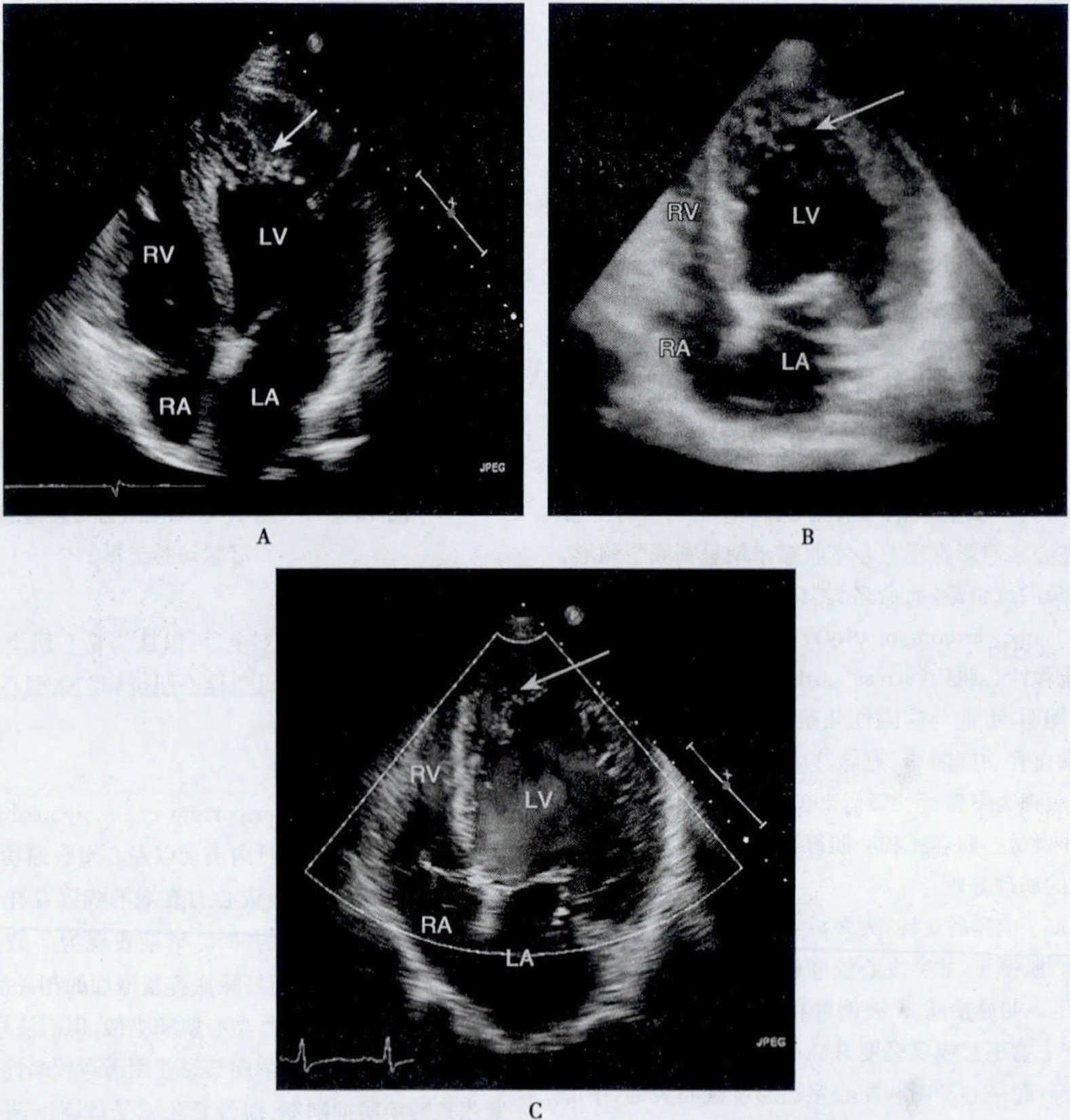
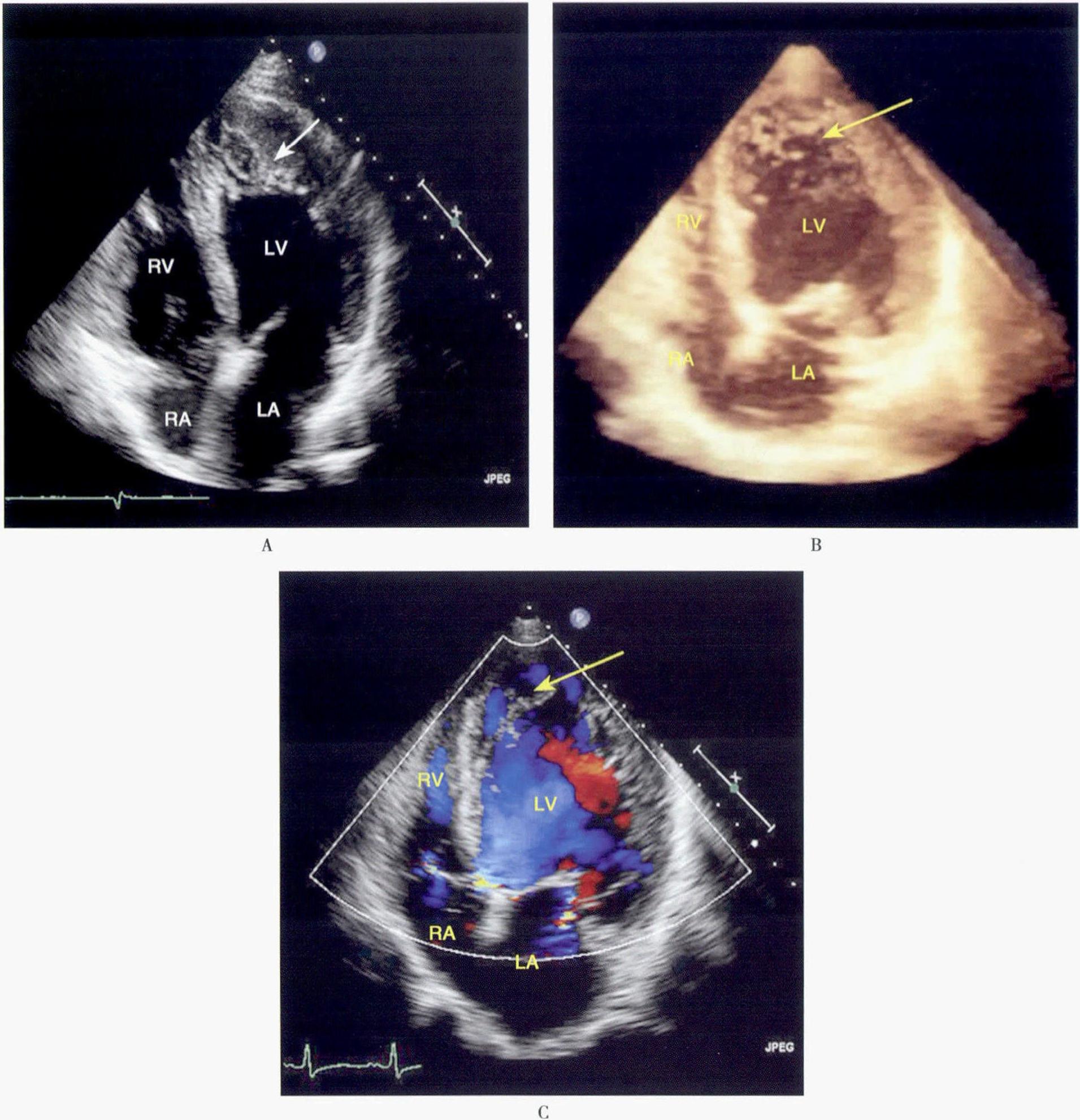
正常胚胎发育的第1个月,心脏冠状动脉循环形成前,胚胎心肌是由海绵状心肌组成,心腔的血液通过其间的隐窝供应相应区域的心肌。胚胎发育5~6周,心室肌逐渐致密化,隐窝压缩成毛细血管,形成冠状动脉微循环系统,致密化过程从心外膜到心内膜,从基底部到心尖部。本病表现为心室肌正常致密化过程停止,形成过多突起肌小梁和深陷的小梁间隙。本病可以是孤立的心脏病变,称为“孤立性心室肌致密化不全”。“心肌窦状隙持续状态”则常用来描述并发于复杂的发绀型先天性心脏病、左心室或右心室梗阻性病变和冠状动脉先天畸形患者的。继发性心肌致密化不全为压力负荷过重和心肌缺血阻止正常胚胎心肌窦隙的闭合所致。但也有人认为此种深陷的间隙衬以内皮细胞,并与心内膜相延续,因此它并非心肌内的窦状隙。目前尚未明确孤立性和继发性心室肌致密化不全是否为同一种疾病。
患者心脏扩大、心肌重量增加、冠状动脉通畅。受累的心室腔内见多发、异常粗大的肌小梁和交错深陷的隐窝,病变可不同程度地累及心室壁的内2/3,肥大肌束的细胞核异形,纤维组织主要出现在心内膜下,其间可见炎症细胞浸润。外层致密心肌厚度变薄,肌束行走及形态学基本正常,细胞核大小均匀。
二、病理生理
(一) 心室收缩和舒张功能不全 舒张功能不全可能是由于异常的心室肌松弛和心腔内过多肌小梁产生心室充盈受限的联合作用所致。过多突起的肌小梁由于血流供需间的不匹配,产生慢性心肌缺血可能是发生进行性收缩功能不全的原因。
(二) 心律失常 可能与肌束极其不规则的分支和连接,等容收缩时室壁张力增加,局部的冠状动脉灌注减低引起组织损伤和激动延迟等潜在的致心律失常原因有关。
(三)体循环栓塞 这可能由于心房颤动和深陷隐窝中的缓慢血流引起血栓形成、栓子脱落发生血栓栓塞而造成的。尸检中曾报道在肌小梁间隙内有血栓形成。