您现在的位置:首页>克罗伊茨费尔特-雅各布病
-
+ 全部展开 -全部收缩
-
-概述
疾病概述:克罗伊茨费尔特-雅各布病(Creutzfeldt-Jakob disease,CJD)简称克雅病,又称为亚急性海绵状脑病(subacute spongiform encephalopathy),是人类最常见的朊蛋白病,主要累及大脑灰质、基底核和脊髓,又称皮质-纹状体-脊髓变性(corticostriatospinal degeneration)。本病由Creutzfeldt(1920)首先报告1例酷似多发性硬化的剖检病例,1921—1923年Jakob报道5例类似的病例,Spielmayer(1923)命名为Creutzfeldt-Jakob病。1929年称为早老性皮质纹状体变性,1940年称为皮质-纹状体-脊髓变性。患者多为中老年人,平均发病年龄60岁,临床主要表现进行性痴呆、肌阵挛、锥体束或锥体外系损伤,数月至1年左右死亡。我国CJD的研究始于20世纪80年代,中山医科大学(1980)首先报告2例经剖检证实的CJD,目前报告的CJD病例已遍及全国。按世界卫生组织要求,国家卫生部及国家疾病预防控制中心决定自2003年起在全国范围内对此病进行监测,也是对非法定传染病唯一实行监测的疾病。
-
-预防
预防:CJD预防已引起WHO及国际医学界高度重视,欧美的疯牛病可通过食物链向全世界扩散,应严格防止病牛肉传入。CJD患者能否直接导致人类传染尚无定论,医务人员及可能接触CJD患者的人应有防护意识,皮肤破损时不能接触患者或实验材料。CJD患者脑活检器械应设标志,用后应高压消毒或用5.25%次氯酸钠液浸泡60分钟以上。CJD患者用过的检查器械应煮沸130℃1小时或苯扎溴铵(新洁尔灭)浸泡1小时以上,活检标本、材料、注射器材和敷料应烧毁。
-
+流行病学
流行病学:本病呈全球性分布,发病率为1/100万。
-
+病因
Prusiner提出,CJD系由特殊的具有感染性蛋白质即朊蛋白(PrP)所致,否定了多年前Gajdusek的非寻常慢病毒感染学说。PrP是一种单基因编码的糖蛋白,由253个氨基酸组成(鼠为254个氨基酸),位于人类20号染色体短臂,可译框架由一个外显子组成,在N末端附近的脯氨酸和甘氨酸短肽有5次重复。正常神经细胞表面也存在朊蛋白PrPc,分子量30~33kD,空间构象主要为α-螺旋结构,蛋白酶K可以溶解。
-
+发病机制
发病机制:1.异常PrPsc与PrPc截然不同,分子量为27~30kD,空间构象约40%为β层状折叠。PrPsc数次集结形成直径为10~20nm、长100~200nm的物质,该物质可能是早期发现的羊瘙痒病相关原纤维(scrapie-associated-fiber,SAF)和朊蛋白微脂粒(prion liposome)。PrPsc不能被蛋白酶K消化而大量地沉积于脑内,破坏中枢神经系统,导致大脑广泛神经细胞凋亡与脱失,形成海绵状脑病。2.本病致病性朊蛋白根据结构不同分为四种亚型,1型与2型存在于散发性CJD,3型为医源性CJD,4型是CJD变异型(vCJD),不同类型CJD发生机制不同。(1) 散发性CJD:发病机制不清,可能是PrP构象异常所致。(2) 医源性CJD(iatrogenic CJD:iCJD):亦称为获得性朊蛋白病(acquired prion disease)。PrPsc污染的组织或器械在脑深部电极检查、颅脑手术、角膜或硬脑膜移植时,以及反复肌注垂体提取的生长激素或性激素而传递感染,经长达数年至数十年的复制发病。手术室与病理实验室工作人员及制备脑源性生物制品者应提高警惕,医务人员应避免身体破损处、结膜和皮肤与患者血液、CSF或组织接触。(3) 家族性CJD:约占15%。Kireshbaun(1924)首先报告经病理证实三代6例CJD患者,Master(1981)复习家族性CJD文献,认为家族性CJD是常染色体显性遗传海绵状脑病。发病年龄通常早于散发性CJD,潜伏期1~40年,同一家系的CJD患者几乎均死于同一年龄段。与散发性CJD不同,家族性CJD除遗传外,均伴PrP基因突变,生成大量PrPsc导致CNS变性。家族性CJD已知可伴178、200、210号密码子突变,178号密码子突变导致FFI或家族性CJD,关键取决于等位基因中129号密码子多态性,178号密码子若与甲硫氨酸结合发生FFI,若与缬氨酸结合发生家族性CJD。特异性基因突变决定朊蛋白病临床类型,并有一定规律可寻(表3-5-7)。目前仍不清楚PrPc如何变为异常的PrPsc。表3-5-7 常见PrP基因突变规律
密码子 碱基 氨基酸 限制酶 51~91 插入性突变144,168,216碱基对 48,56,72残基 102 CCG→CTG CCA→CTA GCA→GTG ATG or GTG TAT→TAG GAC→AAC GTC→ATC TTC→TCC GAG→AAG CAG→CGG ATG→AGG Pro→Leu Pro→Leu Ala→Val Met or Val Tyr→Stop Asp→Asn Val→Ile Phe→Ser Glu→Lys Gln-Arg Met→Arg Dde Ⅰ 引自Tateishi J. British Medical Bulletin,1993,49 (4) :971(4) 变异型CJD(vCJD):是食用被疯牛病感染的牛肉引起的。患者脑组织的动物传染实验证实,与疯牛病有相似的种系特异性。Western印迹分析显示,变异型CJD患者脑组织匀浆(经蛋白酶K消化)分析谱与疯牛病相似性超过了与散发性CJD的相似性。自1996年英国政府承认疯牛病可传染人类后,至2005年初已相继发现暴露于疯牛病病原所致变异型CJD(vCJD)患者180例,已有105例死于vCJD。3.大多数CJD患者无遗传性,也无PrP基因突变。PrPsc沿突触结构广泛沉积于大脑皮质内,称为野生型CJD。129号密码子(codon129)中含甲硫氨酸或缬氨酸比例在东方人与西方人间差异颇大,这种多态性对CJD发病似无直接关系。若178号密码子编码的天门冬氨酸变成天门冬酰胺,呈现丘脑型CJD或FFI;129号密码子由甲硫氨酸变为缬氨酸,患者可有长达20年病史,呈Alzheimer型痴呆;180号密码子的缬氨酸被亮氨酸置换,200号密码子的谷氨酸被赖氨酸置换,232号密码子的甲硫氨酸被精氨酸置换可表现家族性CJD。- +临床表现
临床表现:1. 散发性CJD(sCLD) 是最常见的类型。发病年龄50~75岁,最小14岁,最年长86岁。病程短,平均6个月,进展快,仅14%超过1年,5%超过2年,我国有1例sCJD长达3年。(1) 基本特征为快速进展性痴呆伴神经受损症状,如肌阵挛、锥体束征或锥体外系体征。可有忧郁、幻觉等精神症状,最终发展为无动性缄默。痴呆进展非常迅速,甚至每周或每日都有不同。肌阵挛是sCJD最常见的症状,可因强光、声响或碰触诱发;初期限于某些肌肉,进展后身体各处均可发生。但应注意肌阵挛也见于阿尔茨海默病、皮质基底核变性等。(2) 本病有10%呈卒中样突然发病,数周内死亡。部分患者发病前可有持续数周或数月的非特异前驱症状,如疲劳、头痛、情绪低沉、体重减轻等。2.变异型CJD(vCJD) 临床表现共济失调与行为改变,未发现肌阵挛和特征性脑电图改变,病程较其他类型CJD长,可持续22个月。新变异型CJD(nvCJD)的临床特征是:大多数vCJD在40岁以前发病;病程早期以行为改变为主,散发型CJD早期表现痴呆;EEG缺乏三相慢波,大多数散发型CJD(sCLD)病例有此慢波;神经病理学检查多数病例脑组织斑块针对PrP染色(+),但在sCLD罕见。3. 医源性CJD(iCJD) 可包括iCJD、vCJD和Kuru病。我国仅香港报道1例vCJD,Kuru病在我国从未被发现。至2006年文献报道iCJD400余例,约200例为人体硬脑膜移植所致,其中50%以上发生在日本,我国尚未发现由硬脑膜移植所致的iCJD。此组病例的共同特点是,病情进展缓慢,EEG不呈现周期性同步放电或晚期可出现,PrPSC呈空泡周围性沉积。日本硬脑膜移植所致的iCJD基因检测均为129M/M,52%为斑块性沉积。4.遗传性CLD(gCJD) 2011年德国报道7例E196K突变所引起的gCJD,结合近期文献报道的4例,发现该组病例有若干特殊性。发病年龄63~80岁,平均病程6.5个月。以精神障碍起病并贯穿整个病程,如抑郁、易激惹、行为不适当等。随之出现运动症状,共济失调不明显。脑电图呈现非特异性慢波。30%有阵发性三相波,无周期性。脑脊液14-3-3蛋白均呈阳性,约90%的tau蛋白增高。MRI仅有30%大脑灰质或基底核呈高信号,值得注意的是相当一部分患者大脑白质有改变,尤其年龄70岁以下者更明显。病理改变颇似sCJD,部分可见大脑白质髓鞘脱失。由于该组病例家族无类似病史,若不做基因频谱分析容易误诊为sCJD。- +并发症
- +实验室检查
实验室检查:血常规及生化检查无特异改变。脑脊液基本正常,11%的病例CSF细胞数轻微增高,为淋巴细胞,蛋白质轻度增高。(1) CSF中14-3-3脑蛋白:敏感率为96%,特异性为80%;对进行性痴呆、近期无脑梗死患者的特异性可高达99%,提示测定14-3-3脑蛋白对CJD有很高的诊断价值。14-3-3脑蛋白分子量约30 000,在人类是一种具有多个亚体的正常神经蛋白,它在CJD发病中的作用尚不清楚。一般认为,CJD脑组织大量神经元破坏导致14-3-3脑蛋白释出进入脑脊液,因此14-3-3脑蛋白可作为临床诊断可疑CJD的重要指标,尤其EEG未发现特异变化周期性同步放电(periodic synchronous discharge,PSD)的患者更有意义。应注意一氧化碳中毒、病毒性脑炎、脑梗死及副肿瘤综合征等也可出现14-3-3脑蛋白阳性反应。(2) CSF中130脑蛋白与133脑蛋白:分子量均为30 000,氨基酸序列与14-3-3脑蛋白相同,对CJD也有诊断价值,但其在CSF中含量极低,检测方法较复杂。(3) 血清或CSF-S100蛋白:CJD患者S100蛋白随病情进展呈持续性增高。脑组织中S100蛋白主要存在于星形胶质细胞内,是酸性钙结合蛋白,为两个亚基α、β的同或异二聚体,分子量分别为10.4kD及10.5kD。近来采用免疫荧光法检测224例疑诊CJD患者与35例无痴呆患者血清特异性S100蛋白,其中65例经病理诊断为肯定CJD,6例有遗传基因突变也为肯定CJD,43例为很可能CJD,36例为可能CJD,肯定CJD与很可能CJD患者血清S100蛋白中位数浓度为395pg/ml(s 387pg/ml),显著高于其他疾病(中位数浓度109pg/ml,s 177pg/ml)。以213pg/ml为S100蛋白增高,CJD诊断特异性为81.1%,敏感性77.8%。需注意此项检测的特异性较低,蛛网膜下腔出血、脑卒中、脑缺氧、脑膜炎和多发性硬化等均可能引起血清S100蛋白一过性增高。此种检查也可用于无症状牛海绵状脑病诊断。- +其他辅助检查
其他辅助检查:1.脑电图检查 是CJD临床诊断重要根据,初期仅为广泛非特异性慢波,两侧半球有若干差别;极期呈特异性周期性同步放电(PSD),表现间歇性或连续性中至高波幅尖慢波或棘慢波同步放电,每隔0.6~1.0秒发放1次,持续数秒至十余秒不等。CJD患者PSD不受外界影响,注射地西泮后PSD可明显抑制,晚期PSD消失。PSD与肌阵挛关系密切,伴肌阵挛者PSD出现率为79%。疾病中晚期出现间隔0.5~2秒周期性棘-慢复合波也有诊断价值。脑电图呈阵发性同步放电是诊断sCJD主要依据,但仅在极期出现,晚期则消失,敏感度约65%,特异度为80%,假阳性见于阿尔茨海默病、Lewy体痴呆及血管性痴呆等。2.影像学检查 CJD急性发病或病程较短的病例脑CT检查可完全正常,病程较长可见不同程度脑萎缩,严重者伴脑室扩大。MRI显示双侧尾状核、壳核T2WI呈对称性均质高信号,很少波及苍白球,无增强效应,T1WI可完全正常,具有诊断意义(图3-5-9)。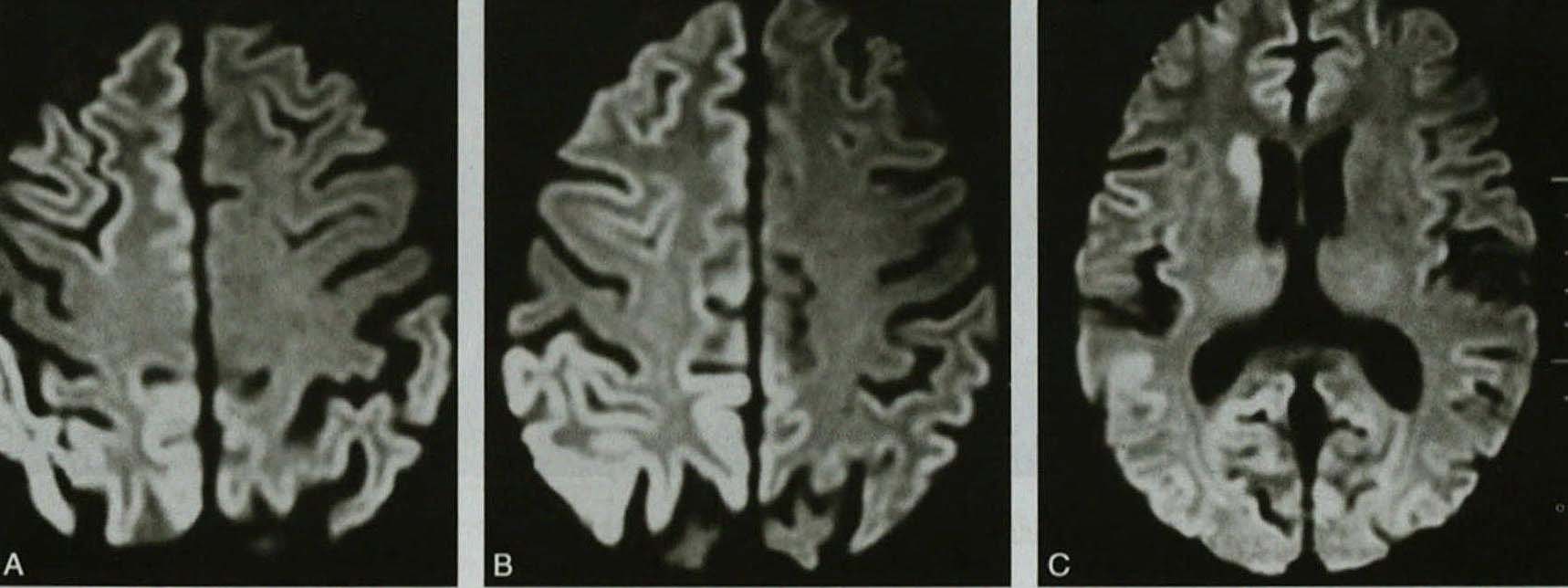
 图3-5-9 CJD患者的脑MRI表现Gertz等(1988)报告1例55岁女患于发病后9个月死亡,死前4周MRI检查发现底节区T2WI高信号。剖检证实整个大脑皮质海绵状变性、胶质增生和轻微神经细胞丧失,尾状核、壳核、苍白球及丘脑病变重于大脑皮质。Pearl等(1989)报告1例68岁女性发病后6个月死亡,死前4个月MRI检查,T2WI见双侧尾状核高信号,剖检证实双尾状核、壳核明显海绵状变性、胶质增生与神经细胞丧失,苍白球、丘脑与大脑皮质病变较轻。也有人报告发病后2个月MRI正常,大脑活检证实CJD,因此MRI正常不能排除CJD。Schroter(2000)采用MRI检查162例临床确诊或可疑sCJD患者,在T2WI及质子密度像109例(67.3%)显示双尾状核与豆状核对称性高信号,在58例其他原因的痴呆患者对照组中仅4例(7%)出现这种改变。磁共振弥散加权像(DWI)远比普通MRI能更早地发现改变,sCJD最常见的改变是颞枕叶灰质对称或不对称的高信号。即使额叶受累,中央前回往往不被涉及,这种中央前回幸免征(precentral sparing sign)可以帮助理解为何sCJD无明显瘫痪。sCJD还可有大脑灰质下白质的高信号,但ADC图像为低信号,此对鉴别快速进展性痴呆的其他疾病很有意义。3.病理 大体所见与病程长短有关。CJD通常于病后12个月内死亡,也可短至数周或迟至数年或更长。病程短者大体病理基本正常,病程长者脑重减轻,有报告1例病程9年的CJD剖检脑重仅575g。脑沟变宽、脑回变窄、脑皮质及基底核萎缩、脑室对称性扩大,脑干、小脑、脊髓外观基本正常。CJD脑萎缩特点是对称性大脑萎缩,极严重病例纹状体、丘脑萎缩,大脑白质通常正常。镜下可见神经元丢失、星形胶质细胞增生、细胞浆空泡形成,感染组织内异常PrP淀粉样斑块,无炎症反应。①海绵状变性:见于大脑皮质深层神经细胞或胶质细胞周围,很少在神经细胞内,呈多数的圆形、椭圆形、不规则形小空泡,可互相融合,直径1~2μm或50μm,严重者见于纹状体、脑干及小脑,急性发病及进展迅速的海绵状变性病变严重;②大脑皮质神经细胞弥散性脱失:第3、5层细胞明显,枕叶尤突出,丘脑和基底核较重,小脑颗粒层细胞脱失较重,脊髓前角细胞染色质溶解或凝集,前角细胞脱失不明显;③胶质细胞增生:急性或慢性进行性病例胶质细胞增生十分突出,星形胶质细胞为主,病程长者更明显;④白质改变:病程较长的CJD脑白质、视神经有轻微改变,脊髓后根神经节、周围神经与自主神经正常;⑤淀粉样斑块:见于小脑分子层,以及齿状核、顶叶、海马、杏仁核、Goll核、三叉神经脊束核与脊髓后角,淀粉斑块见于病程长于15个月的散发性CJD及家族性CJD,短于6个月者无此斑块。电镜典型改变是神经细胞突起终末端及突触间隙不清,界膜空泡(membrane bound vacuole)及突触小泡明显减少。神经细胞浆内染色体减少或消失,脂褐素堆积、髓鞘变薄、轴浆空化和星形胶质细胞增生,胞浆内可见大量次级溶酶体,偶见Rosenthal纤维。淀粉样斑块由7~10nm放射状物质组成,混有10~100nm浓染颗粒,斑块内偶见散在的坏变的神经细胞突起和星形胶质细胞突起。免疫组化染色可用于朊蛋白病诊断,以PrP抗血清为第一抗体,证实PrP在CNS存在与分布,可与其他原因痴呆鉴别。Kitamoto和Tateishi(1988)比较30例CJD、11例GSS及51例非朊蛋白病所致痴呆的免疫组化染色脑组织切片,CJD阳性率为59.0%,GSS为100.0%,其他变性疾病如Alzheimer病、进行性核上麻痹、Huntington舞蹈病、脊髓小脑性共济失调、Pick病等均为阴性。根据PrP存在部位与形式可以区分临床表现不同的斑块型(plaquetype)、突触型(synaptic-type)及周围空泡型(perivacuolar-type)。近年来发现的新变异型CJD(new variant CJD,nvCJD),除了经典的CJD病变,可见大脑与小脑轻微海绵状变性,丘脑和底节改变比大脑灰质重;PrP沉积广泛,枕叶视皮质显著;免疫组化结果与CJD常见的突触型相反,呈斑块型分布。至2001年9月WHO统计欧洲已发现近百例变异型CJD。Heidenhain型以枕叶病变为主,若Brownell及Oppenheimer型以小脑改变为主。
图3-5-9 CJD患者的脑MRI表现Gertz等(1988)报告1例55岁女患于发病后9个月死亡,死前4周MRI检查发现底节区T2WI高信号。剖检证实整个大脑皮质海绵状变性、胶质增生和轻微神经细胞丧失,尾状核、壳核、苍白球及丘脑病变重于大脑皮质。Pearl等(1989)报告1例68岁女性发病后6个月死亡,死前4个月MRI检查,T2WI见双侧尾状核高信号,剖检证实双尾状核、壳核明显海绵状变性、胶质增生与神经细胞丧失,苍白球、丘脑与大脑皮质病变较轻。也有人报告发病后2个月MRI正常,大脑活检证实CJD,因此MRI正常不能排除CJD。Schroter(2000)采用MRI检查162例临床确诊或可疑sCJD患者,在T2WI及质子密度像109例(67.3%)显示双尾状核与豆状核对称性高信号,在58例其他原因的痴呆患者对照组中仅4例(7%)出现这种改变。磁共振弥散加权像(DWI)远比普通MRI能更早地发现改变,sCJD最常见的改变是颞枕叶灰质对称或不对称的高信号。即使额叶受累,中央前回往往不被涉及,这种中央前回幸免征(precentral sparing sign)可以帮助理解为何sCJD无明显瘫痪。sCJD还可有大脑灰质下白质的高信号,但ADC图像为低信号,此对鉴别快速进展性痴呆的其他疾病很有意义。3.病理 大体所见与病程长短有关。CJD通常于病后12个月内死亡,也可短至数周或迟至数年或更长。病程短者大体病理基本正常,病程长者脑重减轻,有报告1例病程9年的CJD剖检脑重仅575g。脑沟变宽、脑回变窄、脑皮质及基底核萎缩、脑室对称性扩大,脑干、小脑、脊髓外观基本正常。CJD脑萎缩特点是对称性大脑萎缩,极严重病例纹状体、丘脑萎缩,大脑白质通常正常。镜下可见神经元丢失、星形胶质细胞增生、细胞浆空泡形成,感染组织内异常PrP淀粉样斑块,无炎症反应。①海绵状变性:见于大脑皮质深层神经细胞或胶质细胞周围,很少在神经细胞内,呈多数的圆形、椭圆形、不规则形小空泡,可互相融合,直径1~2μm或50μm,严重者见于纹状体、脑干及小脑,急性发病及进展迅速的海绵状变性病变严重;②大脑皮质神经细胞弥散性脱失:第3、5层细胞明显,枕叶尤突出,丘脑和基底核较重,小脑颗粒层细胞脱失较重,脊髓前角细胞染色质溶解或凝集,前角细胞脱失不明显;③胶质细胞增生:急性或慢性进行性病例胶质细胞增生十分突出,星形胶质细胞为主,病程长者更明显;④白质改变:病程较长的CJD脑白质、视神经有轻微改变,脊髓后根神经节、周围神经与自主神经正常;⑤淀粉样斑块:见于小脑分子层,以及齿状核、顶叶、海马、杏仁核、Goll核、三叉神经脊束核与脊髓后角,淀粉斑块见于病程长于15个月的散发性CJD及家族性CJD,短于6个月者无此斑块。电镜典型改变是神经细胞突起终末端及突触间隙不清,界膜空泡(membrane bound vacuole)及突触小泡明显减少。神经细胞浆内染色体减少或消失,脂褐素堆积、髓鞘变薄、轴浆空化和星形胶质细胞增生,胞浆内可见大量次级溶酶体,偶见Rosenthal纤维。淀粉样斑块由7~10nm放射状物质组成,混有10~100nm浓染颗粒,斑块内偶见散在的坏变的神经细胞突起和星形胶质细胞突起。免疫组化染色可用于朊蛋白病诊断,以PrP抗血清为第一抗体,证实PrP在CNS存在与分布,可与其他原因痴呆鉴别。Kitamoto和Tateishi(1988)比较30例CJD、11例GSS及51例非朊蛋白病所致痴呆的免疫组化染色脑组织切片,CJD阳性率为59.0%,GSS为100.0%,其他变性疾病如Alzheimer病、进行性核上麻痹、Huntington舞蹈病、脊髓小脑性共济失调、Pick病等均为阴性。根据PrP存在部位与形式可以区分临床表现不同的斑块型(plaquetype)、突触型(synaptic-type)及周围空泡型(perivacuolar-type)。近年来发现的新变异型CJD(new variant CJD,nvCJD),除了经典的CJD病变,可见大脑与小脑轻微海绵状变性,丘脑和底节改变比大脑灰质重;PrP沉积广泛,枕叶视皮质显著;免疫组化结果与CJD常见的突触型相反,呈斑块型分布。至2001年9月WHO统计欧洲已发现近百例变异型CJD。Heidenhain型以枕叶病变为主,若Brownell及Oppenheimer型以小脑改变为主。- +诊断
诊断:CJD早期诊断较困难,可参照以下标准:①中年以上,通常在老年前期或老年期发病;②隐袭起病,缓慢进展,早期智能障碍与行为异常突出,可有痫性发作、皮质盲、肌强直与特征性肌阵挛等;病情迅速进展可数周或数月发展为进行性痴呆、无动性缄默或去皮质强直;③脑电图初期为非特异性慢波,极期可出现周期性同步放电;④脑脊液检出14-3-3脑蛋白,血清证实S100蛋白增高;⑤MRI检查显示双侧尾状核、壳核T2WI对称性均质高信号,苍白球很少波及,无增强效应,DWI常见颞枕叶灰质对称或不对称高信号;⑥脑活检发现大脑灰质海绵状变性、神经细胞缺失、星形胶质细胞增生和PrPsc等。CJD诊断通常采用以下三条标准:①患者在2年内发生进行性痴呆;②出现肌阵挛、视力障碍、小脑症状、无动性缄默等4项症状中2项;③脑电图显示周期性同步放电的特征性改变。患者具备以上三项可诊断很可能的(probable)CJD;仅具备①②项,不具备③项可诊断可能的(possible)CJD;脑活检发现海绵状态和PrPsc为确诊的CJD。- +鉴别诊断
鉴别诊断:CJD需注意与Alzheimer病、进行性核上性麻痹、橄榄脑桥小脑萎缩、脑囊虫病、肌阵挛性癫痫、桥本脑病、颅内转移瘤等鉴别。- +治疗
治疗:目前CJD仍是无法治愈的致死性疾病。已发现缺乏PrPsc基因的鼠不发生CJD,将来有可能应用反义寡核苷酸或基因治疗。对症治疗如痉挛性肌张力增高可用巴氯芬(baclofen,利奥来素),肌阵挛可用氯硝西泮,痴呆可试用茴拉西坦、哌甲酯(利他林)和尼麦角林等。- +预后
约85%的CJD患者发病1年内死亡,少数在发病后3周内或长至8年以上死亡,病程迁延数年者罕见。 - +临床表现