您现在的位置:首页>慢性淋巴细胞白血病
-
+ 全部展开 -全部收缩
-
-概述
疾病概述:慢性淋巴细胞白血病(CLL)是主要发生在中老年人群的一种成熟B淋巴细胞克隆增殖性肿瘤,以淋巴细胞在外周血、骨髓、脾脏和淋巴结聚集为特征。
-
-预防
预防:目前暂无相关资料
-
+流行病学
流行病学:CLL在西方国家是最常见的白血病,在美国其发病率1977年为3.3/10万人,1990年2.3/10万人。发病率近年下降与对CLL和其相关疾病的认识提高,分类进一步完善有关。在西方国家,CLL约占全部成人白血病的30%,男女比例为1.3∶1~2.0∶1,犹太人中CLL发病率较高。CLL在亚洲人较少见。我国对CLL无确切的发病率统计,但CLL占全部成人白血病的比例仅为3%,与日本相似,明显低于西方国家。估计发病率为西方人的1/10,约为0.3/10万人。男女比例为1.3∶1~2.0∶1,细胞遗传学研究不同人种中CLL发病率不同与其具有不同的细胞生物学特性有关。
-
+病因
-
+发病机制
发病机制:CLL的确切发病机制不明,环境因素与CLL发病无明显相关。已报告与其他类型白血病发病有密切相关的因素如电离辐射、化学致癌物、杀虫剂等均与CLL发病无关。病毒感染如HCV(C型肝炎病毒)、EB病毒亦与CLL发病无关。虽然CLL病人中男性明显多于女性,但未发现性激素与CLL发病之间有相关。目前研究集中在CLL发病与遗传因素、染色体、细胞癌基因和抗癌基因改变的关系。1.遗传因素 CLL发病率在白种人和黑种人高,在亚洲黄种人低,其发病率并不因人种的迁居而变化。提示不同种族的某些遗传因素与CLL发病相关。此外,相继有报告在同一个家庭中多人发生B细胞型CLL,CLL患者第一代子女患CLL或其他恶性淋巴增生性疾病的危险性为普通人3倍,且多在年轻时发病,也提示遗传因素在家族性CLL发病中有重要作用,但HLA单一表型与CLL间无明显相关。目前尚未发现与CLL发病的遗传因子,即使单卵双胎子CLL患者,至今未发现有共同的基因异常表现。2.染色体 CLL的细胞遗传学研究较困难,因其淋巴细胞不易受有丝分裂原刺激而增生,不易得到分裂象细胞,近年来,通过改进刺激CLL细胞分裂技术,应用染色体R显带和原位杂交(FISH)法提高了CLL染色体研究成功率。约50%CLL患者发现有克隆染色体异常,而其余正常核型患者可能是正常T细胞核型而未检测到CLL的B细胞异常核型。(1)13号染色体异常:近50%CLL患者有13号染色体长臂缺失。缺失部位多在13q12.3和13q14.3。13q12.3部位缺失,其缺失部位有乳腺癌易感基因(BRCA2)。在13q14.3部位缺失,缺失部位可影响到抑癌基因RB-1(视网膜母细胞基因),DBM(与阻止淋巴细胞恶变有关),LEV1,LEV2和LEV5(与CLL发病有关)。(2)12号染色体异常:12号染色体三体型异常在CLL,初期很少检测到,多在CLL临床病情进展或转为淋巴瘤(Richter综合征)时发现伴有12号染色体三体型的CLL细胞多有复杂型改变及不典型或幼淋细胞形态。提示三体12染色体异常与CLL病情恶化有关。12染色体三体型作用机制可能是通过对位于12q13和12q22之间的某些基因如mdm基因的影响而体现。(3)11号染色体异常:近10%~20%CLL患者有11号染色体移位或缺失,伴有11号染色体异常者临床发病年龄较轻(<55岁),病程常表现为侵袭性。11号染色体异常可累及11q13,目前已认识到此部位包括肿瘤抑制基因-MEN-1(多发性内分泌肿瘤综合征Ⅰ型)。最常见的11号染色体缺失在11q14-24之间,特别在11q22.3-23.1之间,在此部位中可能有肿瘤抑制基因RDX(多发性神经纤维瘤Ⅱ型肿瘤抑制基因同类物)和AIM(遗传性共济失调-毛细胞血管扩张症突变基因),这两种基因的功能与激活肿瘤抑制基因p53有关。p53基因具有调节细胞周期和维持基因稳定作用,其表达产物可使异常细胞进入细胞周期时被阻滞在S1期,便于异常细胞有更多的时间进行DNA修复,如细胞不能自行修复受损的DNA,则会自行凋亡。(4)6号染色体异常:包括6号染色体短臂及长臂异常。6号染色体短臂异常目前尚未发现有相应特定基因功能改变。6q21-q24异常患者临床常表现为幼淋细胞增多和侵袭性病程。此外,TNF-α(肿瘤坏死因子α),和LY-α(淋巴a)其基因均位于6号染色体长臂,此两种因子与促进CLL细胞增生,抑制正常淋巴细胞和骨髓细胞增生有关。(5)14号染色体异常:常表现为易位。在CLL患者中少见,在淋巴瘤患者中多见t(11;14)(q13;q32)易位:在CLL中罕见。14 q32含有免疫球蛋白a重链同型开关基因,而11q13有细胞周期素D1基因(cyclic D1)t(11;14),常见于外套型非霍奇金淋巴瘤。t(14:18)CLL患者罕见,常见于低度恶性滤泡型淋巴瘤。3.特殊基因改变(1)p53基因:p53基因为一种重要的肿瘤抑制基因,位于17p13.1部位,编码53-kD核酸磷酸蛋白。其突变或缺陷可能为近半数肿瘤患者的致病原因。17号染色体短臂缺失仅见于10%~15%的CLL患者。此外,还有10%~15% CLL患者有p53基因突变,伴有p53基因突变患者多为进展型,具有白血病细胞高增生率,生存期短,对一线治疗药物抵抗的临床特点,见于半数Richter综合征和B细胞幼淋细胞白血病,提示p53基因突变可能是某些CLL患者病程中获得性改变。(2)多剂耐药基因(MDR):约40%CLL患者MDR-1基因表达增高,MDR-1位于7q21.1,编码170kD跨膜部糖蛋白。在CLL患者B细胞中MDR-1表达增加而在正常B细胞中表达不增加,此外由于治疗或其他因素也可诱导MDR-1基因表达增加,MDR基因异常表达更多是促进CLL患者病程进展原因而不是CLL原发病因。(3)bcl-2:bcl-2基因位于染色体18q21,大多数CLL患者由于bcl-2基因重排而表达增加。约有5%左右CLL患者bcl-2基因重排是位于2号和8号染色体上的IGk或λ轻链基因与位于18号染色体bcl基因易位。但除基因重排外,CLL白血病细胞bcl-2表达增加与其基因位点的低甲基化有关。可能还有一些尚未了解的基因亦参与作用,使CLL细胞抵抗凋亡。4.细胞因子 CLL细胞具有分泌多种细胞因子的能力,如TNF-α,TGF-β (转移生长因子β)、IL-7(白介素-7)、IL-5、IL-2等,这些因子具有直接或间接刺激CLL白血病细胞增生或防止CLL细胞凋亡作用,同时具有抑制正常淋巴细胞和骨髓造血有关细胞增生作用,因而细胞因子与CLL患者发病和疾病进展均相关。细胞动力学研究显示,CLL患者周围血中被3H标记的白细胞数量少,提示大多数白细胞处于休止期,(G0期)而不增殖,同时发现几乎所有的CLL的白细胞均表达高水平的抗凋亡蛋白bcl-2,及低水平的凋亡收蛋白bax,故bcl-2/bax比例失衡,致细胞凋亡受阻,符合临床上大量成熟小淋巴细胞积聚的现象,构成CLL的主要病理基础。
-
+临床表现
临床表现:本病多见于50岁以上患者,男女比例约为2:1。起病缓慢,多无自觉症状。许多患者在常规体检或因其他疾病就诊时才被发现。有症状者早期可表现为乏力、疲倦,而后出现食欲减退、消瘦、低热、盗汗等。60%~80%的患者有淋巴结肿大,多见于头颈部、锁骨上、腋窝及腹股沟。肿大淋巴结一般为无痛性,中等硬度,无粘连,随病程进展可逐渐增大或融合。CT扫描可发现纵隔、腹膜后、肠系膜淋巴结肿大。肿大的淋巴结可压迫气管、上腔静脉、胆道或输尿管而出现相应症状。半数以上患者有轻至中度的脾大,肝大多为轻度,胸骨压痛少见。晚期患者可出现贫血、血小板减少和粒细胞减少,常易并发感染。由于免疫功能失调,常并发自身免疫性疾病,如自身免疫性溶血性贫血(AIHA)、免疫性血小板减少性紫癜(ITP)等。部分患者可转化为幼淋巴细胞白血病(PLL)、Richter综合征(转化为弥漫大B细胞淋巴瘤等),或继发第二肿瘤。
-
+并发症
并发症:1.感染 CLL患者死亡和病情恶化的主要原因之一是感染,可累及约40%的患者。低γ球蛋白血症是感染和病情恶化的主要原因之一。此外,还有粒细胞缺乏、T细胞功能异常等。最常见的是细菌感染,病毒感染(尤其是疱疹病毒感染)约占15%,真菌感染较少见。2.继发肿瘤 9%~20%的CLL患者可继发第2肿瘤,最常见的继发肿瘤为软组织肉瘤、肺癌等。CLL患者发生多发性骨髓瘤的可能性比常人增加10倍,但二者并非起源于同一恶性B细胞克隆。CLL继发急性髓细胞白血病的危险并不增加。
-
+实验室检查
实验室检查:以淋巴细胞持续性增多为主要特征。白细胞>10×109/L,淋巴细胞比例≥50%,淋巴细胞绝对值≥5×109/L(至少持续3个月)。大多数患者的白血病细胞形态与成熟小淋巴细胞类同,胞质少,胞核染色质呈凝块状。少数患者细胞形态异常,胞体较大,不成熟,胞核有深切迹(Reider细胞)。偶可见原始淋巴细胞。中性粒细胞比值降低。随病情进展,可出现血小板减少和贫血。有核细胞增生明显活跃或极度活跃,淋巴细胞≥40%,以成熟淋巴细胞为主。红系、粒系及巨核系细胞增生受抑,至晚期可明显减少。伴有溶血时,幼红细胞可代偿性增生。淋巴细胞具有单克隆性,呈现B细胞免疫表型特征。细胞膜表面免疫球蛋白(sIg)为弱阳性表达,多为IgM或IgM和IgD型,呈κ或λ单克隆轻链型;小鼠玫瑰花结试验阳性;CD5、CD19、CD79α、CD23阳性;CD20、CD22、CD11c弱阳性;FMC7、CD79β阴性或弱阳性;CD10、cyclinD1阴性。CLL缺乏特异性标记,可应用免疫表型的积分系统来进行鉴别。患者中60%有低γ球蛋白血症,20%抗人球蛋白试验阳性,8%出现AIHA。常规显带1/3~1/2的患者有克隆性核型异常。由于CLL细胞有丝分裂相较少,染色体异常检出率低,间期荧光原位杂交(FISH)技术能明显提高检出率,可检测到>80%的患者存在染色体异常。如13q14缺失(50%)、12号染色体三体(20%)、11q22~23缺失、17p13缺失和6q缺失等。单纯13q14缺失提示预后良好,12号染色体三体和正常核型预后中等,17p13及11q22~23缺失预后差。50%~60%的CLL发生免疫球蛋白重链可变区(IgVH)基因体细胞突变,IgVH突变发生于经历了抗原选择的记忆B细胞(后生发中心),此类病例生存期长;无IgVH突变者,起源于未经抗原选择的原始B细胞(前生发中心)。无IgVH突变的CLL细胞多数高表达CD38、ZAP70,均与不良预后相关。约10%~15%的CLL存在p53基因突变(该基因位于17p13),与疾病进展有关,对治疗有抵抗,生存期短。
-
+其他辅助检查
其他辅助检查:根据临床表现、症状、体征,可选择做CT、X线、B超等检查。
-
+诊断
诊断:达到以下3项标准可以诊断:①外周血B淋巴细胞(CD19+细胞)计数≥5×109/L;B 淋巴细胞<5×109/L时,如存在CLL细胞骨髓浸润所致的血细胞减少,也可诊断CLL。②外周血涂片中特征性的表现为小的、形态成熟的淋巴细胞显著增多,其细胞质少、核致密、核仁不明显、染色质部分聚集,并易见涂抹细胞。外周血淋巴细胞中不典型淋巴细胞及幼稚淋巴细胞≤55%。③典型的免疫表型:CD19+、CD5+、CD23+、CD10-、FMC7-、CD43+/-、CCND1-;表面免疫球蛋白(sIg)、CD20及CD79b弱表达(dim)。流式细胞学确认B细胞的克隆性,即B细胞表面限制性表达κ或λ轻链(κ∶λ>3∶1或<0.3∶1)或>25%的B细胞sIg不表达。
-
+鉴别诊断
鉴别诊断:具体鉴别诊断从如下几方面进行:1. CLL:典型的CLL在涂片上一般包括三类细胞(外周血涂片优于骨髓涂片):①成熟小淋巴细胞;②中等大小带有明显核仁的淋巴细胞(副免疫母细胞或者幼淋细胞)(比例<55%);③涂抹细胞。骨髓活检可见间质、结节或弥漫性浸润,细胞核小、圆形,染色质呈颗粒状。2. MCL:细胞中等大小,核边缘明显不规则或有切迹,类似于生发中心的中心细胞。少数形态学亚型类似原始细胞或者多形细胞,必须与PLL、急性淋巴细胞白血病(ALL)鉴别。极少数形态学类似CLL细胞,甚至免疫表型为CD5+CD23+,故cyclin D1阳性或t(11;14)至关重要。3. SMZL:成熟小淋巴细胞,无核仁,具有特征性的极性绒毛。骨髓活检可见结节样的间质性浸润,该特点有助于排除HCL。4. HCL:相对于其他B-CLPD,HCL 的诊断更依赖于免疫表型,尤其是FCM检查。细胞表面有绒毛状突起,细胞中等大小,染色质略显疏松,核仁缺少或模糊,大量浅蓝色胞质,呈现为特征性的煎鸡蛋样。骨髓穿刺常为“干抽”。骨髓活检显示间质浸润,大面积的弥漫性骨髓侵犯少见,网硬蛋白纤维可增加。5. 脾B细胞淋巴瘤/白血病,不能分类:HCL-V细胞有明显的核仁和曲核,但缺乏毛状细胞外观形。SDRPSBCL 细胞常呈绒毛状细胞外形,常累及骨髓窦状隙和外周血。6. B-PLL:细胞中等大小,胞质量少呈淡蓝色,有一个明显的核仁。骨髓侵犯以间质或结节样浸润为主。形态学与CLL的幼淋巴细胞转化、MCL母细胞变异型区分困难,需要依赖于免疫分型和细胞遗传学。7. FL:小淋巴细胞,伴有裂的细胞核。骨髓活检可见诊断性的形态学特征:骨小梁旁浸润。8. LPL/WM:由小淋巴细胞、浆细胞样淋巴细胞和浆细胞组成,经常可见增多的肥大细胞。部分胞质内(Russell 小体)或者细胞核内(Dutcher 小体)的PAS 阳性的球形包涵体。骨髓活检可见间质、结节或弥漫性浸润,偶见小梁旁聚集。1. CLL:特点为CD5和CD23与CD19共表达,但CD20dim(dim:弱表达)和sIgdim,FMC7、CD22和CD79b常阴性或弱表达,不表达cyclin D1[免疫组织化学(IHC)]与CD10。可根据英国马斯登皇家医院(Royal Marsden Hospital,RMH)免疫标志积分与其他B-CLPD 鉴别(表1),CLL 4~5 分,其他B-CLPD 0~2 分, 积分3 分时建议进行FISH 检测除外MCL,CD200 在CLL和HCL细胞中高表达,而在其他B-CLPD(包括MCL、FL和SMZL)中表达阴性或低表达。
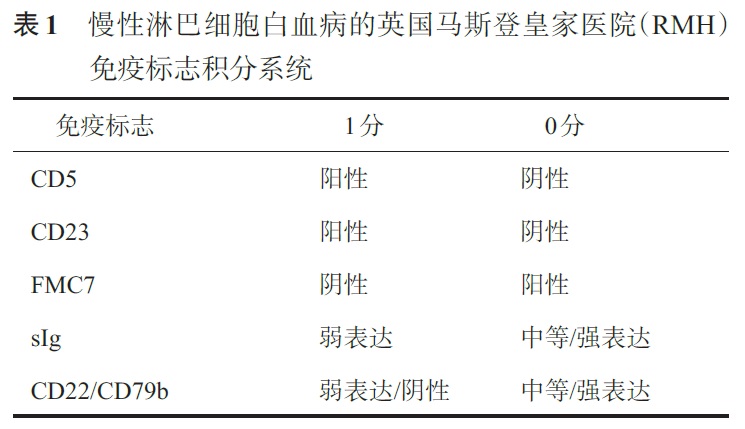 2. MCL:表达成熟B 细胞相关抗原,同时表达CD5 和cyclin D1,CD10、CD23(25% 弱阳性)和Bcl- 6 常阴性。CD20、CD79b和sIg 表达比CLL强,且CD23阴性和FMC7阳性,可以与CLL相鉴别。此外,CD11c 在MCL常阴性,也有助于与CLL相鉴别(1/3 的CLL患者CD11c 阳性)。3. SMZL:表达成熟B细胞相关抗原,但无特异性抗原表达。CD5、CD23 和CD10 阴性,采用CLL 免疫积分标准<2分,CD79b、FMC7 和sIg 表达强度明显高于CLL。CD5 和CD23 阴性可与CLL 鉴别;cyclin D1 和CD5 阴性可与MCL鉴别;CD103和Annexin A1(IHC)阴性可与HCL鉴别;CD10和Bcl-6(IHC)阴性可与FL鉴别。4. HCL:表达成熟B细胞相关抗原,且CD20bright(bright:强阳性)和CD22bright。HCL 细胞还表达CD11cbrigh、CD25brigh、CD103 和CD123,FMC7 和sIg 阳性,Annexin A1(IHC)在HCL特异性表达。CD5、CD10、CD23和CD43阴性。5. 脾B 细胞淋巴瘤/白血病,不能分类:HCL-V表达成熟B 细胞相关抗原,CD11c 和FMC7 阳性,CD103 阳性或阴性,但CD25、CD123 和Annexin A1(IHC)阴性。SDRPSBCL也表达成熟B 细胞相关抗原,但CD11c、CD25、CD103、CD123和Annexin A1(IHC)常为阴性。6. B-PLL:表达成熟B细胞相关抗原,FMC7 阳性,CD5和CD23大多阴性,少数CD5和CD23阳性,CD11c、CD25和CD103阴性。7. FL:表达成熟B细胞相关抗原,生发中心抗原CD10、Bcl-2(IHC)和Bcl-6(IHC)阳性,部分患者FMC7和CD23 阳性。8. LPL/WM:表达成熟B 细胞相关抗原,同时CD38 和CD138 阳性,肿瘤细胞表面和一些细胞质中有免疫球蛋白,通常IgM型,也可IgG型,不表达IgD。1. CLL:由于CLL细胞为相对成熟的淋巴细胞,分裂能力差,常规核型分析难以获得中期分裂象,间期FISH是最常用的细胞遗传学检测技术,采用FISH 与一组探针可以发现大约80%的CLL患者存在细胞遗传学异常。常见的遗传学异常包括del(13q14)、+ 12、del(11q22.3)、del(17p13)、del(6q23)等。2. MCL:t(11;14)是特征性的染色体异常。FISH 是检测t(11;14)的理想技术(敏感性为80%~100%),常规细胞遗传学检测t(11;14)敏感性为50%~75%,PCR 的敏感性仅为30%~50%。极少数患者t(11;14)阴性。3. MZL:无特异性遗传学异常。SMZL常见的遗传学异常包括del(7q21-32)和+3;NMZL和结外MALT型MZL常见的遗传学异常包括+3和t(11;18)(q21;q21)。4. HCL:无特异性遗传学异常,但多数HCL 患者存在BRAF V600E 突变,可用于与其他B-CLPD(包括HCL-V)鉴别。5. 脾B 细胞淋巴瘤/白血病,不能分类:HCL-V无特异性遗传学异常,在一些病例证实存包括del(17p13)、14q32 或8q24 易位等复杂核型异常。SDRPSBCL也无特异性遗传学异常,已有发现存在del(17p13)、t(9;14)等遗传学异常。6. B-PLL:无特异性遗传学异常,复杂核型异常常见。常见的遗传学异常包括del(17p13)、del(13q)、+12、del(6q)。7. FL:主要的细胞遗传学异常为t(14;18),由此产生的Bcl-2/IgH 融合基因,见于85%~90% FL。8. LPL/WM:无特异性遗传学异常,常见的遗传学异常包括del(6q21-q23)、del(13q)、+18、+4、del(17p13)、t(9;14)。MYD88 L265P 突变发生率高,可用于LPL/WM 的诊断。具体各种疾病的免疫表型及遗传学特征见表2
2. MCL:表达成熟B 细胞相关抗原,同时表达CD5 和cyclin D1,CD10、CD23(25% 弱阳性)和Bcl- 6 常阴性。CD20、CD79b和sIg 表达比CLL强,且CD23阴性和FMC7阳性,可以与CLL相鉴别。此外,CD11c 在MCL常阴性,也有助于与CLL相鉴别(1/3 的CLL患者CD11c 阳性)。3. SMZL:表达成熟B细胞相关抗原,但无特异性抗原表达。CD5、CD23 和CD10 阴性,采用CLL 免疫积分标准<2分,CD79b、FMC7 和sIg 表达强度明显高于CLL。CD5 和CD23 阴性可与CLL 鉴别;cyclin D1 和CD5 阴性可与MCL鉴别;CD103和Annexin A1(IHC)阴性可与HCL鉴别;CD10和Bcl-6(IHC)阴性可与FL鉴别。4. HCL:表达成熟B细胞相关抗原,且CD20bright(bright:强阳性)和CD22bright。HCL 细胞还表达CD11cbrigh、CD25brigh、CD103 和CD123,FMC7 和sIg 阳性,Annexin A1(IHC)在HCL特异性表达。CD5、CD10、CD23和CD43阴性。5. 脾B 细胞淋巴瘤/白血病,不能分类:HCL-V表达成熟B 细胞相关抗原,CD11c 和FMC7 阳性,CD103 阳性或阴性,但CD25、CD123 和Annexin A1(IHC)阴性。SDRPSBCL也表达成熟B 细胞相关抗原,但CD11c、CD25、CD103、CD123和Annexin A1(IHC)常为阴性。6. B-PLL:表达成熟B细胞相关抗原,FMC7 阳性,CD5和CD23大多阴性,少数CD5和CD23阳性,CD11c、CD25和CD103阴性。7. FL:表达成熟B细胞相关抗原,生发中心抗原CD10、Bcl-2(IHC)和Bcl-6(IHC)阳性,部分患者FMC7和CD23 阳性。8. LPL/WM:表达成熟B 细胞相关抗原,同时CD38 和CD138 阳性,肿瘤细胞表面和一些细胞质中有免疫球蛋白,通常IgM型,也可IgG型,不表达IgD。1. CLL:由于CLL细胞为相对成熟的淋巴细胞,分裂能力差,常规核型分析难以获得中期分裂象,间期FISH是最常用的细胞遗传学检测技术,采用FISH 与一组探针可以发现大约80%的CLL患者存在细胞遗传学异常。常见的遗传学异常包括del(13q14)、+ 12、del(11q22.3)、del(17p13)、del(6q23)等。2. MCL:t(11;14)是特征性的染色体异常。FISH 是检测t(11;14)的理想技术(敏感性为80%~100%),常规细胞遗传学检测t(11;14)敏感性为50%~75%,PCR 的敏感性仅为30%~50%。极少数患者t(11;14)阴性。3. MZL:无特异性遗传学异常。SMZL常见的遗传学异常包括del(7q21-32)和+3;NMZL和结外MALT型MZL常见的遗传学异常包括+3和t(11;18)(q21;q21)。4. HCL:无特异性遗传学异常,但多数HCL 患者存在BRAF V600E 突变,可用于与其他B-CLPD(包括HCL-V)鉴别。5. 脾B 细胞淋巴瘤/白血病,不能分类:HCL-V无特异性遗传学异常,在一些病例证实存包括del(17p13)、14q32 或8q24 易位等复杂核型异常。SDRPSBCL也无特异性遗传学异常,已有发现存在del(17p13)、t(9;14)等遗传学异常。6. B-PLL:无特异性遗传学异常,复杂核型异常常见。常见的遗传学异常包括del(17p13)、del(13q)、+12、del(6q)。7. FL:主要的细胞遗传学异常为t(14;18),由此产生的Bcl-2/IgH 融合基因,见于85%~90% FL。8. LPL/WM:无特异性遗传学异常,常见的遗传学异常包括del(6q21-q23)、del(13q)、+18、+4、del(17p13)、t(9;14)。MYD88 L265P 突变发生率高,可用于LPL/WM 的诊断。具体各种疾病的免疫表型及遗传学特征见表2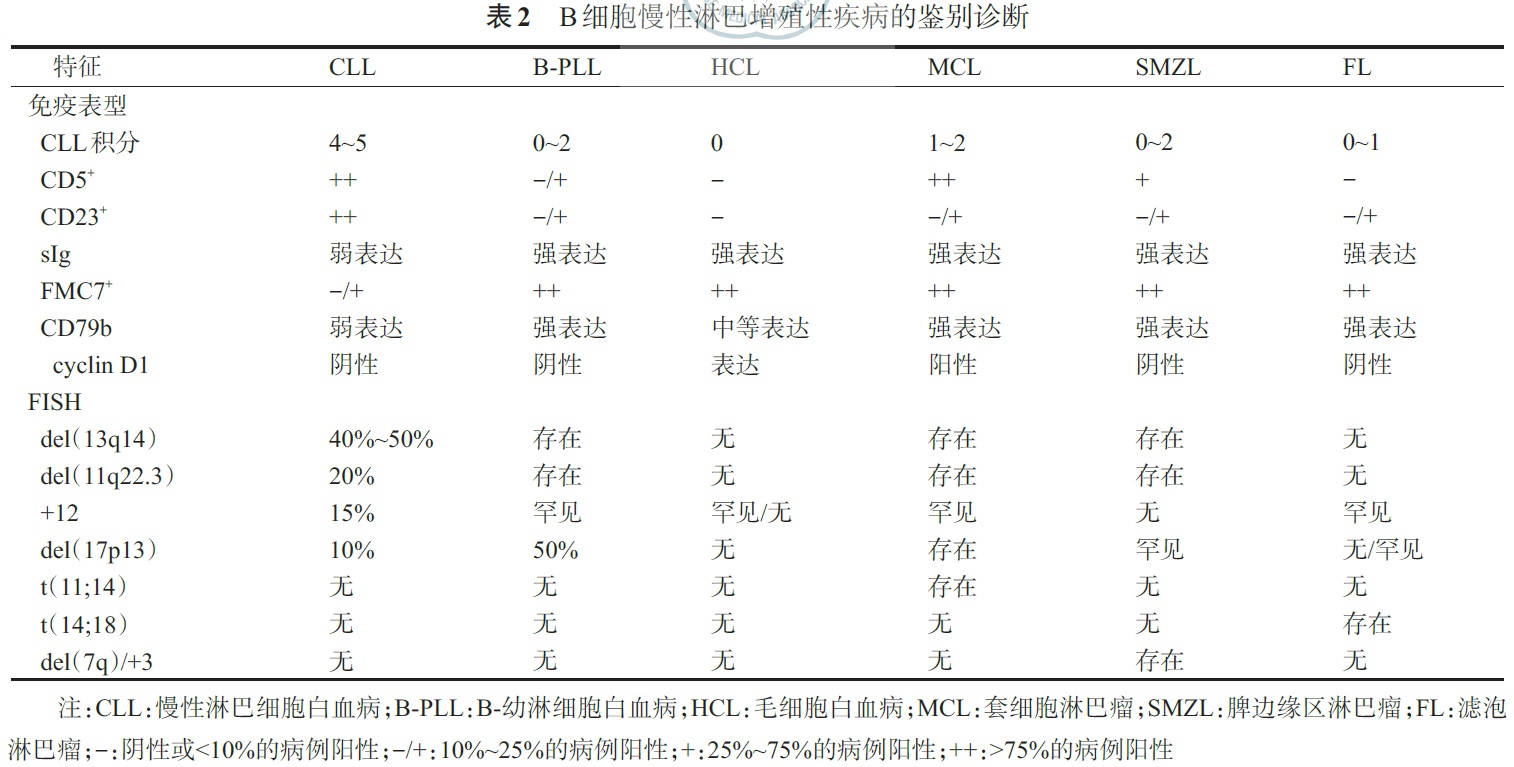
-
+治疗
治疗:不是所有CLL 都需要治疗,只有具备以下至少1项时方可开始治疗。1. 进行性骨髓衰竭的证据:表现为血红蛋白和(或)血小板进行性减少。2. 巨脾(如左肋缘下>6 cm)或进行性或有症状的脾肿大。3. 巨块型淋巴结肿大(如最长直径>10 cm)或进行性或有症状的淋巴结肿大。4. 进行性淋巴细胞增多,如2 个月内淋巴细胞增多>50%,或淋巴细胞倍增时间(LDT)<6 个月。当初始淋巴细胞<30×109/L,不能单凭LDT 作为治疗指征。5. 淋巴细胞计数>200×109/L,或存在白细胞淤滞症状。6. 自身免疫性溶血性贫血(AIHA)和(或)免疫性血小板减少症(ITP)对皮质类固醇或其他标准治疗反应不佳。7. 至少存在下列一种疾病相关症状:①在以前6 个月内无明显原因的体重下降≥10%;②严重疲乏[如ECOG体能状态≥2;不能进行常规活动];③无感染证据,体温>38.0℃,≥2 周;④无感染证据,夜间盗汗>1个月。8. 临床试验:符合所参加临床试验的入组条件。不符合上述治疗指征的患者,每2~6 个月随访1 次,随访内容包括临床症状及体征、肝/脾/淋巴结肿大情况和血常规等。治疗前(包括复发患者治疗前)必须对患者进行全面评估。评估的内容包括:①病史和体格检查:特别是淋巴结(包括咽淋巴环和肝脾大小);②体能状态:ECOG和(或)疾病累积评分表(CIRS)评分;③B症状:盗汗、发热、体重减轻;④血常规检测:包括白细胞计数及分类、血小板计数、血红蛋白等;⑤血清生化检测,包括肝肾功能、电解质、LDH、β2-MG等;⑥骨髓活检±涂片:治疗前、疗效评估及鉴别血细胞减少原因时进行,典型病例的诊断、常规随访无需骨髓检查;⑦常规染色体核型分析(CpG刺激);⑧HBV 检测;⑨有条件的单位尽可能进行FISH 检测del(13q)、+12、del(11q)、del(17p),建议开展分子生物学技术检测p53、IGHV、NOTCH1、SF3B1、BIRC3、MYD88 等基因突变,以帮助判断预后和指导治疗。特殊情况下检测:免疫球蛋白定量;网织红细胞计数和直接抗人球蛋白试验(怀疑有溶血时必做);超声心动图检查(拟采用蒽环类或蒽醌类药物治疗时);颈、胸、腹、盆腔增强CT检查等。选择根据FISH 结果[del(17p)和del(11q)]、年龄及身体状态进行分层治疗。患者的体能状态和实际年龄均为重要的参考因素;治疗前评估患者的伴发疾病(CIRS 评分)和身体适应性极其重要。体能状态良好(包括肌酐清除率≥70 ml/min 及CIRS 评分≤6 分)的患者建议选择一线标准治疗,其他患者则使用减低剂量化疗或支持治疗。1. 无del(17p)/p53 基因突变或del(11q)CLL患者的治疗方案推荐(按优先顺序):(1)存在严重伴随疾病的虚弱患者(不能耐受嘌呤类似物):①苯丁酸氮芥±泼尼松±利妥昔单抗(RTX);②环磷酰胺±泼尼松±RTX;③RTX;④皮质类固醇冲击疗法。(2)≥70 岁或存在严重伴随疾病(CIRS 评分>6分)的<70 岁患者:①苯达莫司汀±RTX;②苯丁酸氮芥± 泼尼松±RTX;③环磷酰胺± 泼尼松±RTX;④RTX;⑤氟达拉滨±RTX;⑥克拉屈滨±RTX。(3)<70 岁且无严重伴随疾病(CIRS 评分≤6分):①氟达拉滨+环磷酰胺±RTX±米托蒽醌(FC±RTX±M);②苯达莫司汀±RTX;③氟达拉滨±RTX;④苯丁酸氮芥±泼尼松±RTX;⑤环磷酰胺±泼尼松±RTX。2. 伴del(17p)/p53 基因突变CLL患者的治疗方案推荐(按优先顺序):(1)目前所有治疗方案疗效不佳,建议参加临床试验。(2)HDMP(大剂量甲泼尼龙)±RTX±新鲜冰冻血浆(FFP)。(3)调整的Hyper-CVAD±RTX。(4)氟达拉滨+环磷酰胺±RTX。(5)氟达拉滨±RTX。(6)苯达莫司汀±RTX。(7)苯丁酸氮芥±泼尼松±RTX。(8)环磷酰胺±泼尼松±RTX。3. 伴del(11q)CLL 患者的治疗方案推荐(按优先顺序):(1)≥70 岁或存在严重伴随疾病(CIRS 评分>6分)的<70 岁患者:①苯达莫司汀±RTX;②苯丁酸氮芥±泼尼松±RTX;③环磷酰胺±泼尼松±RTX;④减低剂量的氟达拉滨+环磷酰胺±RTX;⑤RTX;⑥氟达拉滨±RTX。(2)<70 岁且无严重伴随疾病(CIRS 评分≤6分):①氟达拉滨+环磷酰胺±RTX;②苯达莫司汀±RTX;③氟达拉滨±RTX;④苯丁酸氮芥±泼尼松±RTX;⑤环磷酰胺±泼尼松±RTX。定义:复发:患者达到完全缓解(CR)或部分缓解(PR),≥6 个月后疾病进展(PD);难治:治疗失败(未获CR或PR)或最后1次化疗后<6个月PD。复发、难治患者的治疗指征、治疗前检查同一线治疗,在选择治疗方案时除考虑患者的年龄、体能状态及遗传学等预后因素外,应同时综合考虑患者既往治疗方案的疗效(包括持续缓解时间)及耐受性等因素。1. 无del(17p)/p53 基因突变患者的治疗方案推荐(按优先顺序):(1)持续缓解≥2 年:重复一线治疗方案或选用新方案。(2)持续缓解<2 年:首选一线治疗未用过的治疗方案。≥70 岁或存在严重伴随疾病(CIRS 评分>6 分)的<70 岁患者:①苯达莫司汀±RTX;②减低剂量的氟达拉滨+环磷酰胺±RTX;③HDMP±RTX;④来那度胺/沙利度胺±RTX;⑤剂量密集RTX;⑥新鲜冰冻血浆+RTX;⑦苯丁酸氮芥±泼尼松±RTX;⑧环磷酰胺±泼尼松±RTX。<70 岁且无严重伴随疾病(CIRS 评分≤6 分):①氟达拉滨+环磷酰胺±RTX;②苯达莫司汀±RTX;③HDMP±RTX;④调整的HyperCVAD±RTX;⑤来那度胺/沙利度胺±RTX;⑥OFAR(奥沙利铂+氟达拉滨+阿糖胞苷±RTX);⑦苯丁酸氮芥±泼尼松±RTX;⑧环磷酰胺±泼尼松±RTX。2. 伴del(17p)/p53 基因突变CLL患者的治疗方案推荐(按优先顺序,首选一线治疗未用过的治疗方案):(1)目前所有治疗方案疗效不佳,建议参加临床试验。(2)HDMP±RTX±新鲜冰冻血浆。(3)调整的HyperCVAD±RTX。(4)氟达拉滨+环磷酰胺±RTX。(5)苯达莫司汀±RTX。(6)来那度胺/沙利度胺±RTX。(7)OFAR。(8)苯丁酸氮芥±泼尼松±RTX。(9)环磷酰胺±泼尼松±RTX。目前无标准维持治疗方案,不推荐常规维持治疗,可以进行科学设计的维持治疗探索。近年来欧美国家针对CLL 的治疗药开发获得快速发展,已经上市或即将上市的药物包括阿仑单抗、GA101、奥法木单抗(Ofatumumab)、依鲁替尼、Idelalisib等,如有合适的临床试验,值得积极参加。自体造血干细胞移植有可能改善患者的无进展生存(PFS),但并不延长总生存(OS)期,不推荐常规采用。异基因造血干细胞移植是CLL 的唯一治愈手段,但由于CLL 主要为老年患者,仅少数适合移植。适应证:①氟达拉滨耐药:对以氟达拉滨为基础的治疗无反应或治疗后12 个月内复发;②具有p53 基因异常的患者;③伴del(11q),治疗仅达≤PR的患者;④Richter转化患者。1. Richter 综合征:伴有弥漫大B细胞淋巴瘤/霍奇金淋巴瘤转化的CLL 患者,大多数预后很差,中位生存期大多不超过1 年,治疗建议参照侵袭性淋巴瘤的治疗策略及方案。2. 自身免疫性血细胞减少症:激素是一线治疗。激素无效的患者可选择行静脉注射丙种球蛋白(IVIG)、RTX、环孢素及脾切除等治疗。3. 感染:感染的防治包括:CLL 患者化疗前后病毒、细菌、真菌感染的预防和治疗;乙肝病毒携带者治疗中的预防等。1. CLL患者存在较大感染风险,反复感染的患者IVIG维持IgG≥5 g/L。2. 每年接种流感疫苗、每5 年接种肺炎球菌疫苗,避免所有活疫苗的接种。在CLL患者的治疗中应定期进行疗效评估,诱导治疗通常以6 个疗程为宜,建议治疗3~4 个疗程时进行中期疗效评估,疗效标准见表3。
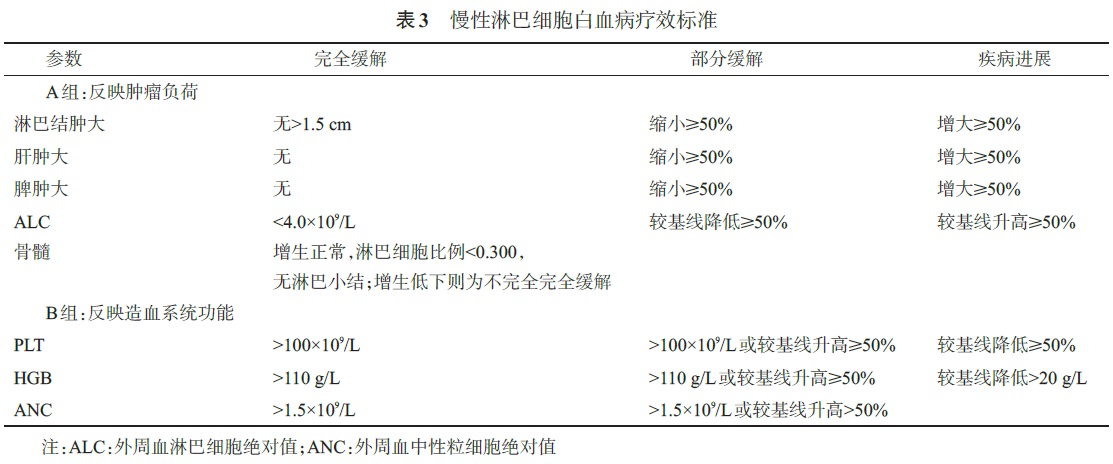 CR:达到表3 所有标准,无疾病相关症状;不完全CR(CRi):除骨髓未恢复正常外,其他符合CR标准;PR:至少达到2 个A组标准+1 个B组标准;疾病稳定(SD):疾病无进展同时不能达到PR;PD:达到任何1 个A 组或B 组标准;复发:患者达到CR 或PR,≥6 个月后PD;难治:治疗失败(未获CR或PR)或最后1 次化疗后<6 个月PD;微小残留病阴性:多色流式细胞术检测残存白血病细胞<1×10-4。燃瘤反应(tumor flare reaction):来那度胺等免疫调节剂治疗后引起的疼痛性淋巴结肿大、淋巴细胞增多、皮疹和骨痛。伴淋巴细胞增高的PR:B细胞受体信号通路的小分子抑制剂如BTK 抑制剂依鲁替尼和PI3Kd 抑制剂Idelalisib 治疗后出现短暂淋巴细胞增高,淋巴结、脾脏缩小。此时单纯的淋巴细胞增高不作为疾病进展标准。完成诱导治疗(一般6 个疗程)达CR或PR的患者,应该定期进行随访,包括每3 个月血细胞计数及肝、脾、淋巴结触诊检查等。应该特别注意免疫性血细胞减少症(AIHA、ITP)、继发恶性肿瘤(包括骨髓增生异常综合征、急性髓系白血病及实体瘤等)的出现。
CR:达到表3 所有标准,无疾病相关症状;不完全CR(CRi):除骨髓未恢复正常外,其他符合CR标准;PR:至少达到2 个A组标准+1 个B组标准;疾病稳定(SD):疾病无进展同时不能达到PR;PD:达到任何1 个A 组或B 组标准;复发:患者达到CR 或PR,≥6 个月后PD;难治:治疗失败(未获CR或PR)或最后1 次化疗后<6 个月PD;微小残留病阴性:多色流式细胞术检测残存白血病细胞<1×10-4。燃瘤反应(tumor flare reaction):来那度胺等免疫调节剂治疗后引起的疼痛性淋巴结肿大、淋巴细胞增多、皮疹和骨痛。伴淋巴细胞增高的PR:B细胞受体信号通路的小分子抑制剂如BTK 抑制剂依鲁替尼和PI3Kd 抑制剂Idelalisib 治疗后出现短暂淋巴细胞增高,淋巴结、脾脏缩小。此时单纯的淋巴细胞增高不作为疾病进展标准。完成诱导治疗(一般6 个疗程)达CR或PR的患者,应该定期进行随访,包括每3 个月血细胞计数及肝、脾、淋巴结触诊检查等。应该特别注意免疫性血细胞减少症(AIHA、ITP)、继发恶性肿瘤(包括骨髓增生异常综合征、急性髓系白血病及实体瘤等)的出现。 -
+预后