您现在的位置:首页>多系统萎缩
-
+ 全部展开 -全部收缩
-
-概述
疾病概述:多系统萎缩(multiple system atrophy,MSA)在1969年被首次提出,对这一疾病曾有不同的命名,如纹状体黑质变性(striatonigral degeneration,SND)、橄榄脑桥小脑萎缩(OPCA)和Shy-Drager综合征。目前认为,MSA是一种散发性神经系统退行性疾病,临床表型主要包括自主神经功能障碍、帕金森综合征、共济失调及锥体系统功能障碍等,主要分为两种临床类型:帕金森型(MSA-parkinsonian,MSA-P)和小脑型(MSA-cerebellar,MSA-C),不同的患者可表现为各种症状的重叠组合。含α-突触核蛋白(α-synuclein)的神经胶质胞质包涵体(glial cytoplasmic inclusion,GCI)是MSA的神经病理学标志性特征(Wakabayashi et al.1998),因此该病被归于突触核蛋白病(synucleinopathy)。
- -预防
-
+流行病学
流行病学:MSA多在50~60岁起病,发病率男性高于女性。欧美人群MSA的年发病率约为0.6/10万,50岁以上人群中为3/10万;不同的研究者报道的患病率为(1.9 ~4.9)/10万,日本的MSA人群患病率约为8/10万(Wenning et al. 2009)。近年来,MSA的发病率在世界范围内呈上升趋势,已受到国内外神经学医师的广泛关注,目前,北美多系统萎缩研究组(NAMSA-SG)、日本多系统萎缩研究学会(JAMSASA)、欧洲多系统萎缩研究组(EMSA-SG)及中国多系统萎缩研究组(CNMSA-SG)等相继已成立,从临床、病因学、发病机制及治疗等多个方面进行研究。
-
+病因
-
+发病机制
发病机制:MSA的神经病理改变见于中枢神经系统的多个部位,如纹状体、黑质致密部、蓝斑、小脑、脑桥核、下橄榄核及中间外侧柱等。特征性病理改变是广泛、密集分布的α-突触核蛋白阳性的神经胶质胞质包涵体(Wenning et al. 2005)(图3-21-2);分布密度与神经变性的程度及病程相关,在神经变性程度较严重的脑区,如壳核、黑质、脑桥核、小脑普肯耶细胞、脊髓中间外侧柱等密度较高,但无显著神经变性的脑区亦可见到α-突触核蛋白阳性的神经胶质胞质包涵体。在MSA患者脑实质还可见反应性胶质细胞增生、小胶质细胞活化、铁质沉积及髓鞘变性等。Jellinger等(2005)提出,依据神经胶质胞质包涵体(GCIs)的分布部位及神经元缺失程度进行MSA的病理分级,可量化评价GCIs的密度及神经变性程度。多系统萎缩患者的神经元亦可见α-突触核蛋白聚集,分布于胞质、胞核及轴突(Yoshida et al. 2007)(图3-21-3)。
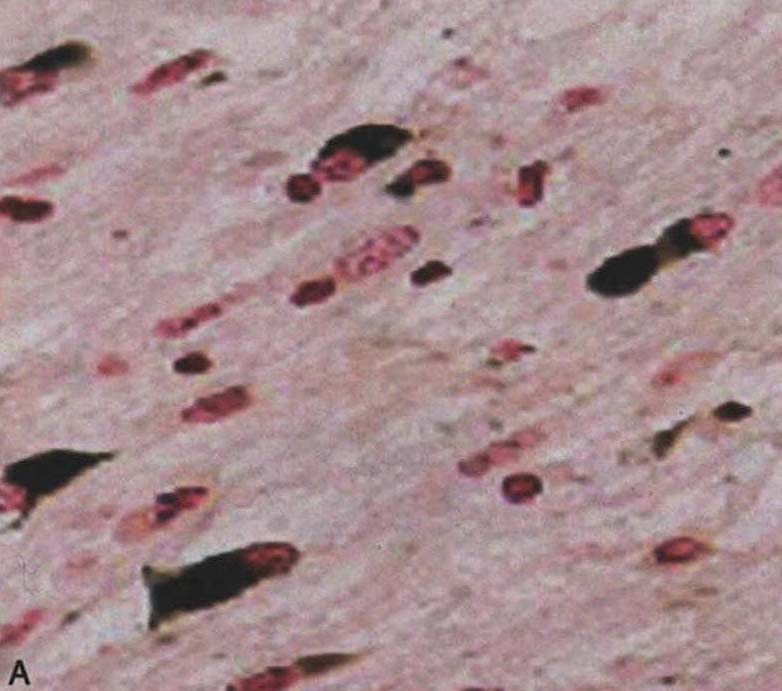
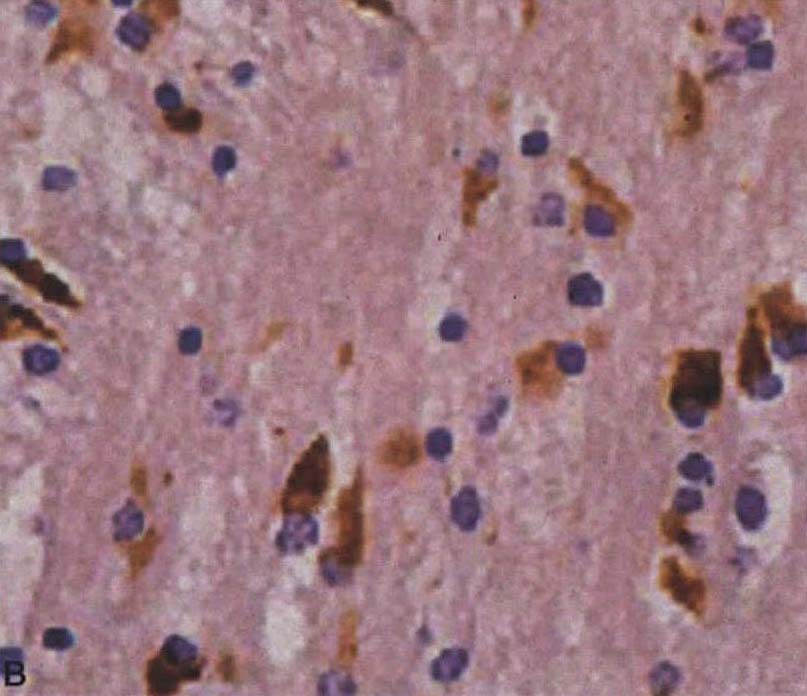
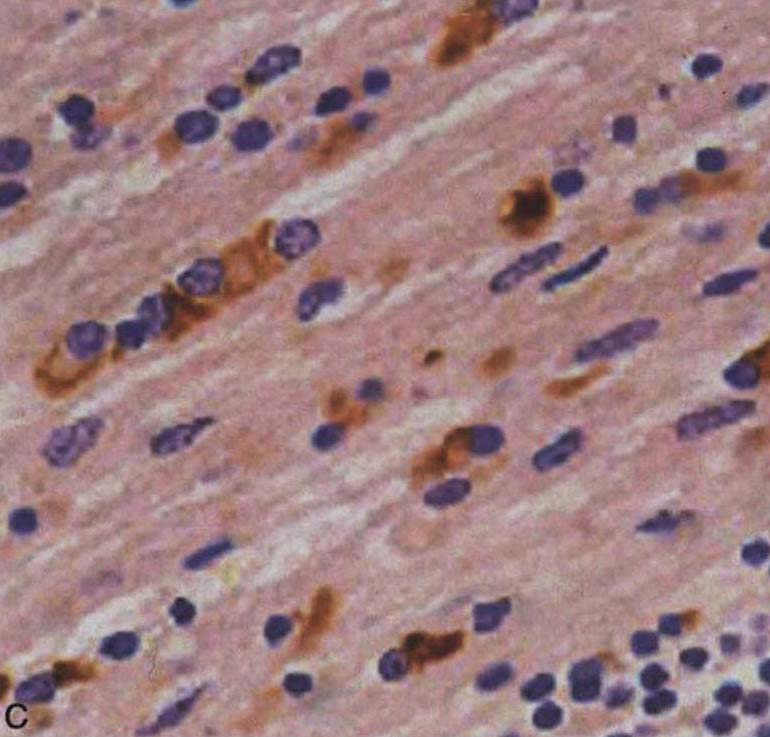
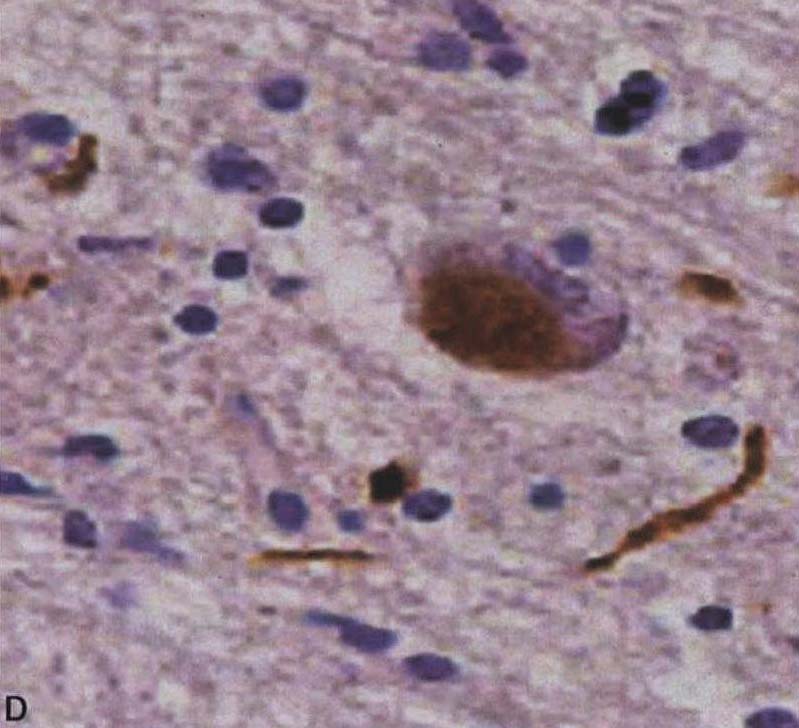 图3-21-2 少突胶质细胞α-突触核蛋白阳性包涵体A.苍白球(银染);B.脑桥基底部(α-突触核蛋白);C.额叶白质(抗泛素);D.脑桥基底部神经细胞胞浆包涵体和神经突(α-突触核蛋白)。A~D×4000(Wenning et al.2005)
图3-21-2 少突胶质细胞α-突触核蛋白阳性包涵体A.苍白球(银染);B.脑桥基底部(α-突触核蛋白);C.额叶白质(抗泛素);D.脑桥基底部神经细胞胞浆包涵体和神经突(α-突触核蛋白)。A~D×4000(Wenning et al.2005)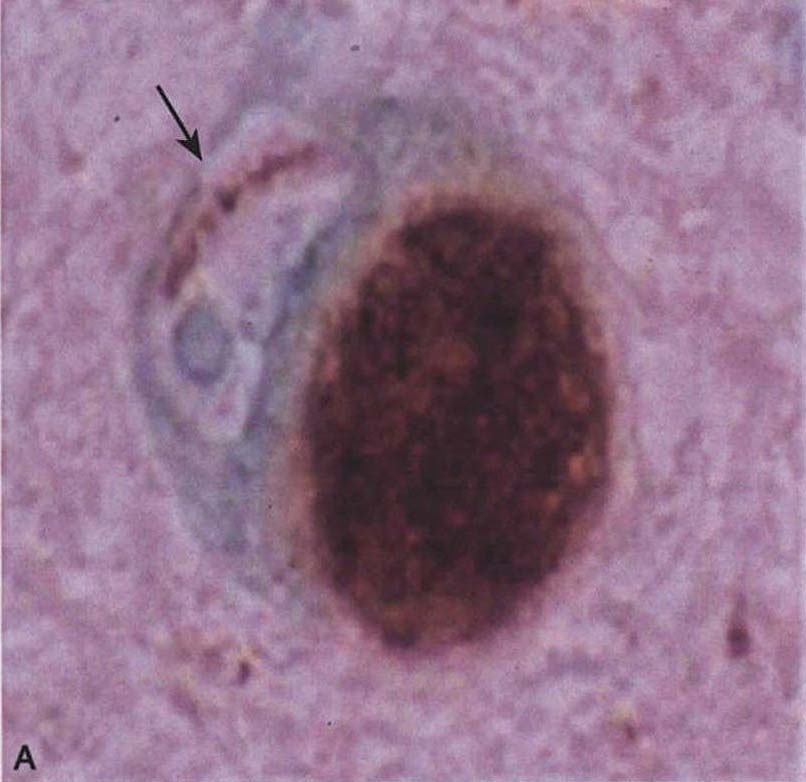
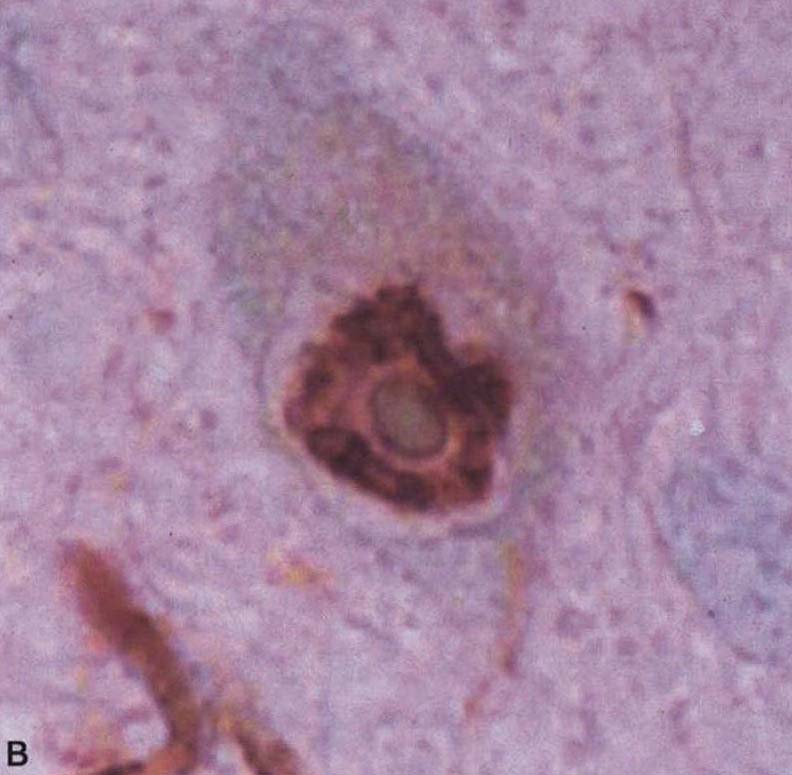
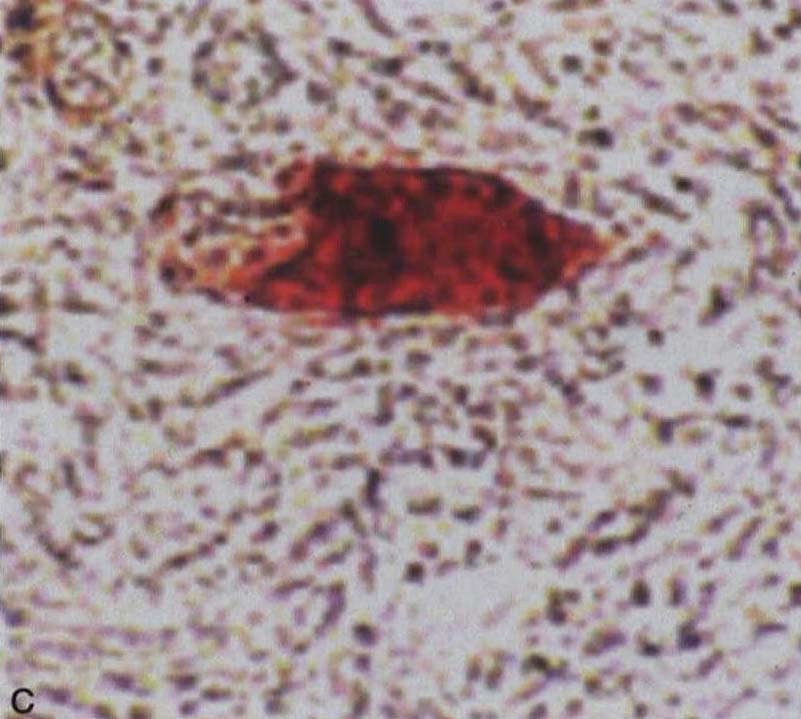
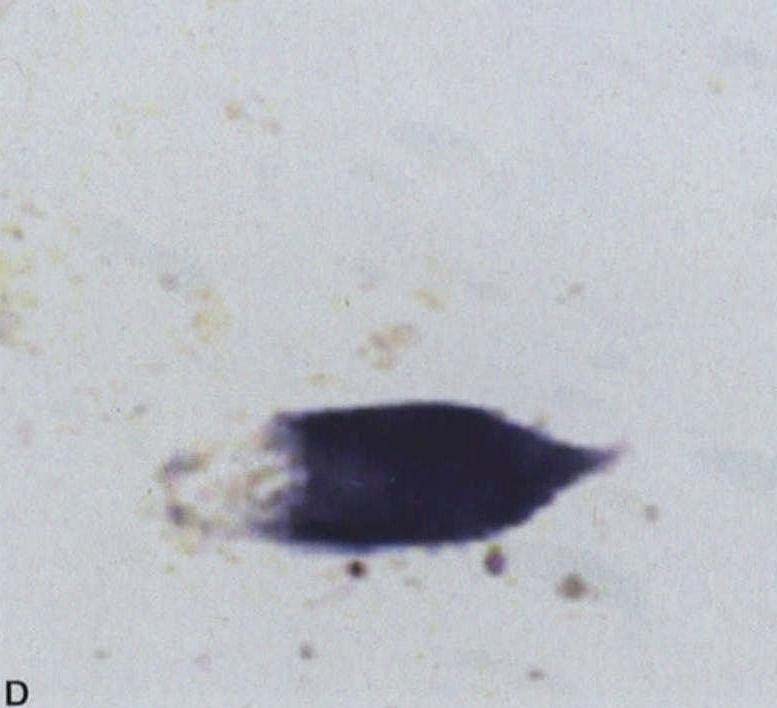 图3-21-3A.脑桥核神经元胞浆α-突触核蛋白阳性包涵体,细胞核内镶边状沉积(箭头所示);B.脑桥核神经细胞核内泛素阳性包涵体;C,D.脑桥基底部神经胶质胞质包涵体,C为泛素阳性,D为14-3-3蛋白阳性。A,B ×550;C,D×600(Wenning et al. 2005)MSA的神经胶质胞质包涵体(GCIs)提示最初的损害在白质,这种胶质细胞的慢性病变可能影响少突胶质细胞与轴索间的营养功能,导致继发性神经元损伤,但这种包涵体代表原发性损伤抑或细胞损伤的非特异性继发特征,目前尚不清楚。除了GCIs外,脑内还出现广泛的髓鞘变性,在MSA发病过程中可能起重要作用(Wenning et al. 2008)。GCIs的成分复杂,包括α-突触核蛋白、泛素、微管蛋白、14-3-3蛋白、tau蛋白和一些少突胶质细胞标志物。少突胶质细胞是α-突触核蛋白异常聚集最早且最严重的细胞,故少突胶质细胞变性可能是神经元变性的触发因素。在少突胶质细胞特异启动子控制的高表达α-突触核蛋白转基因小鼠,少突胶质细胞有过度磷酸化的α-突触核蛋白异常聚集现象,以及神经元变性改变,类似人类MSA的病理学特征(Yazawa et al.2005)。α-突触核蛋白在少突胶质细胞异常聚集的机制仍未阐明。有研究发现,在多系统萎缩早期,少突胶质细胞即有P25α聚集,P25α为少突胶质细胞特异性磷酸化蛋白或称为促微管聚合蛋白(TPPP),与神经髓鞘功能有密切相关(Song et al.2007)。MSA患者发生α-突触核蛋白聚集前,P25α被转移至少突胶质细胞胞体。P25α在体外能够促进α-突触核蛋白聚集(Lindersson et al. 2005),这支持原发性少突胶质细胞病变先于神经元变性,少突胶质细胞变性可能导致神经元髓鞘脱失,激活小胶质细胞及诱发氧化应激,导致神经元变性死亡。
图3-21-3A.脑桥核神经元胞浆α-突触核蛋白阳性包涵体,细胞核内镶边状沉积(箭头所示);B.脑桥核神经细胞核内泛素阳性包涵体;C,D.脑桥基底部神经胶质胞质包涵体,C为泛素阳性,D为14-3-3蛋白阳性。A,B ×550;C,D×600(Wenning et al. 2005)MSA的神经胶质胞质包涵体(GCIs)提示最初的损害在白质,这种胶质细胞的慢性病变可能影响少突胶质细胞与轴索间的营养功能,导致继发性神经元损伤,但这种包涵体代表原发性损伤抑或细胞损伤的非特异性继发特征,目前尚不清楚。除了GCIs外,脑内还出现广泛的髓鞘变性,在MSA发病过程中可能起重要作用(Wenning et al. 2008)。GCIs的成分复杂,包括α-突触核蛋白、泛素、微管蛋白、14-3-3蛋白、tau蛋白和一些少突胶质细胞标志物。少突胶质细胞是α-突触核蛋白异常聚集最早且最严重的细胞,故少突胶质细胞变性可能是神经元变性的触发因素。在少突胶质细胞特异启动子控制的高表达α-突触核蛋白转基因小鼠,少突胶质细胞有过度磷酸化的α-突触核蛋白异常聚集现象,以及神经元变性改变,类似人类MSA的病理学特征(Yazawa et al.2005)。α-突触核蛋白在少突胶质细胞异常聚集的机制仍未阐明。有研究发现,在多系统萎缩早期,少突胶质细胞即有P25α聚集,P25α为少突胶质细胞特异性磷酸化蛋白或称为促微管聚合蛋白(TPPP),与神经髓鞘功能有密切相关(Song et al.2007)。MSA患者发生α-突触核蛋白聚集前,P25α被转移至少突胶质细胞胞体。P25α在体外能够促进α-突触核蛋白聚集(Lindersson et al. 2005),这支持原发性少突胶质细胞病变先于神经元变性,少突胶质细胞变性可能导致神经元髓鞘脱失,激活小胶质细胞及诱发氧化应激,导致神经元变性死亡。 -
+临床表现
临床表现:1. 多系统萎缩的临床表现 主要包括自主神经功能障碍、类帕金森病表现、共济失调及锥体束征等。Köllensperger等(2010)总结欧洲的一项来自10个国家19个研究中心共计437例患者的临床资料显示,MSA患者临床表现类似,平均发病年龄57.8岁,入组时平均病程5.8年。根据诊断标准,68%的患者归于MSA-P,32%的患者归于MSA-C。符合很可能诊断标准的患者占72%,可能的诊断占28%。几乎所有患者均出现自主神经功能障碍,其中排尿障碍占83%;体位性低血压占75%;具有帕金森综合征表现占87%;具有小脑共济失调表现占64%。2. 自主神经功能障碍是MSA各亚型的共同特征 包括:(1) 体位性低血压:常因胸、腰髓侧角节前交感神经元变性引起,卧位血压通常正常,少数病例夜间高血压,站立时血压迅速下降,除早期偶有代偿性心率加快外,一般发作时无心率变化,也无晕厥患者常见的苍白、出汗、恶心等症状。患者感觉站立行走时头晕眼花,平卧时症状改善,日间困倦,餐后尤明显。有些严重患者采用蹲踞缓解头晕,个别患者可出现晕厥,也有个别患者有明显的体位性低血压但自觉症状不明显,测量血压显示收缩压下降>20mmHg或舒张压下降>10mmHg。(2) 泌尿生殖系统功能障碍:是骶髓侧角副交感神经变性所致,主要表现尿频、尿急、尿失禁及夜尿增多,残余尿量增加。女性患者尿失禁更明显;男性患者有尿不净感,常误诊为前列腺肥大,术后无改善。几乎所有的男性患者都有勃起功能障碍。(3)其他如患者常感觉排便无力,但与老年人常见的大便干燥引起的便秘不同;患者多感觉排汗减少,尤其下肢皮肤干燥,严重者夏季可出现体温升高;有些病例晚期可出现夜间喘息性呼吸困难,迷走神经背核受损引起声音嘶哑、吞咽困难,以及心搏骤停导致猝死。3.大多数MSA患者在病程某一阶段出现帕金森综合征,表现运动迟缓、双侧肢体僵直、姿势性震颤、姿势平衡障碍、颅颈部肌张力障碍等,典型搓丸样震颤极少见,姿势不稳出现早且进展较帕金森病迅速。左旋多巴对MSA患者疗效欠佳,约30%的患者在疾病早期左旋多巴有一定的疗效,但难以持久。4. MSA-C型患者最常见行走不稳,常伴肢体共济失调、小脑性构音障碍,进展速度较其他晚发性小脑共济失调快,一般发病5年后丧失行走能力。锥体系损害可见腱反射亢进、病理征及假性延髓麻痹等,少数患者出现下运动神经元损害,引起肌萎缩。5.最新研究显示,颈项前驱、吸气性喘鸣及眼动异常在MSA具有早期诊断价值(Köllensperger et al.2010,Geser et al. 2006)。大多数MSA患者起病前数年出现快速眼动期行为障碍(REM behavior disorder,RBD),可能是MSA的前驱症状(Iranzo et al. 2005),郝红琳等(2010)对66例PD患者、30例MSA患者及65名性别、年龄匹配的健康对照进行睡眠状况调查及夜间多导睡眠监测,结果显示,RBD在PD和MSA患者中出现率明显增高,部分RBD发生先于变性病。
- +并发症
-
+实验室检查
实验室检查:生化检测 24小时尿去甲肾上腺素及肾上腺素排泄减少,放射性核素标记显示去甲肾上腺素代谢正常,提示患者在正常生理状态下不能释放儿茶酚胺。直立时血浆肾素释放减少,有些患者醛固酮分泌亦减少,肾素-醛固酮系统障碍可能与钠贮存不足有关。个别患者出现低钠血症。
-
+其他辅助检查
其他辅助检查:1.神经影像学 脑MRI检查在多系统萎缩诊断中有重要价值,可显示MSA患者壳核、小脑中脚及脑干萎缩,T2WI可见脑桥“十字征”,壳核“裂隙征”及壳核背外侧低信号,但这些改变缺乏特异性。Horimoto等(2002)根据不同病程阶段T2WI异常信号的特点提出脑桥“十字征”和壳核“裂隙征”的分期方法(表3-21-2)。表3-21-2 不同病程T2 WI异常信号的脑桥“十字征”及壳核“裂隙征”的分期法
脑桥“十字征”可分成六期 夏程等(2007)对11例多系统萎缩的临床分型和影像学改变特点进行分析,我们研究组对143例符合Gilman诊断标准的MSA患者进行临床分型和诊断分级,根据Horimoto分期对108例有明显神经影像改变的患者的脑桥“十字征”和壳核“裂隙征”进行分期(图3-21-4)(杨斯柳et al. 2010)。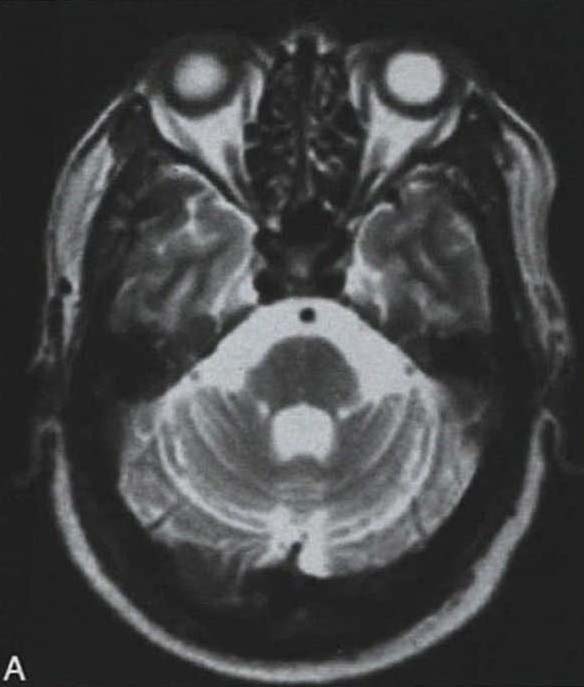
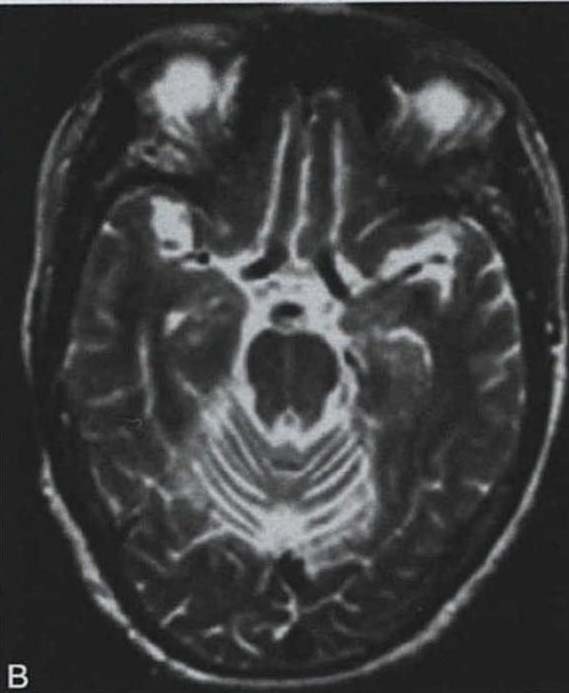
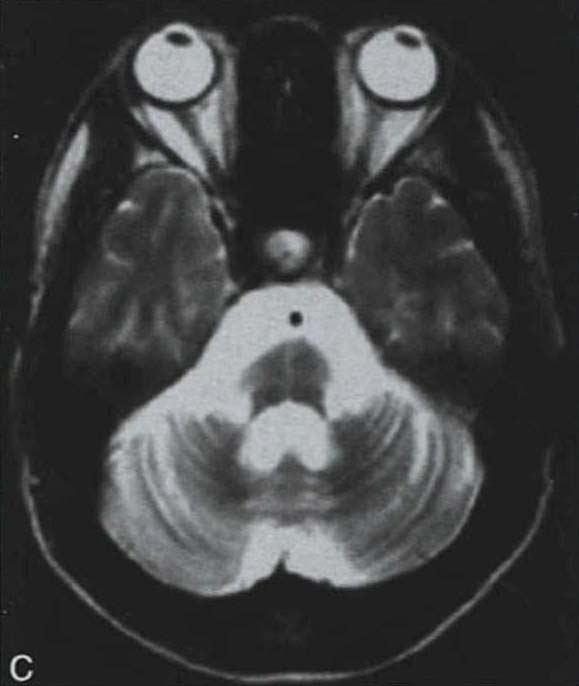
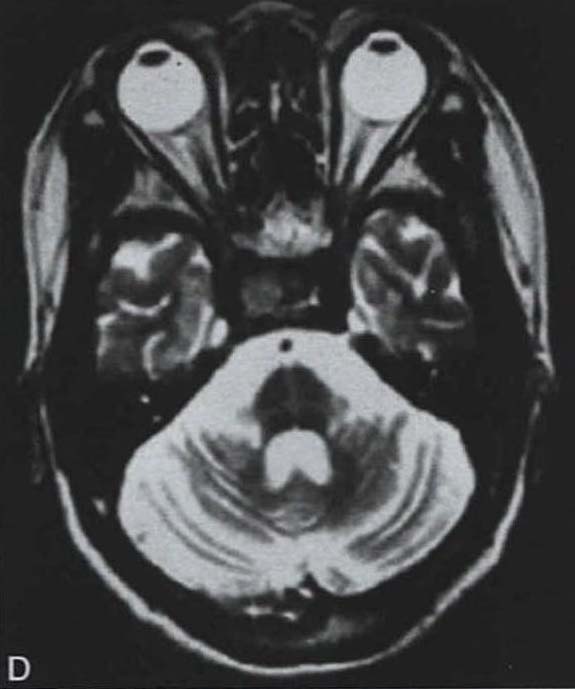
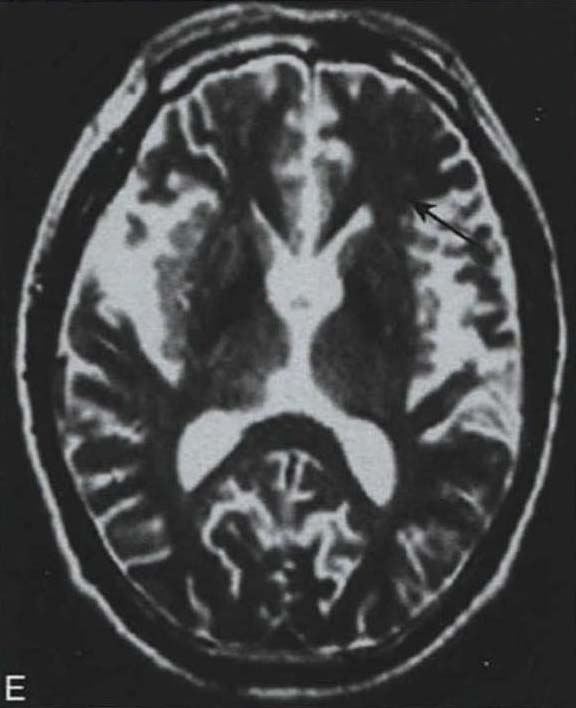
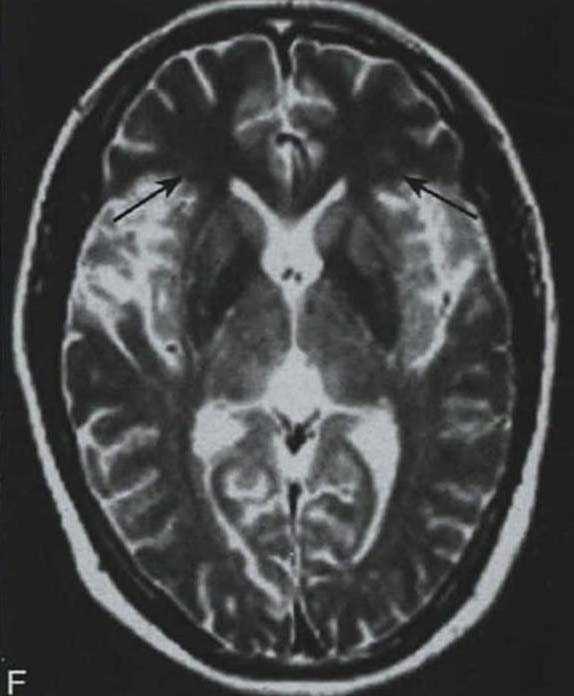
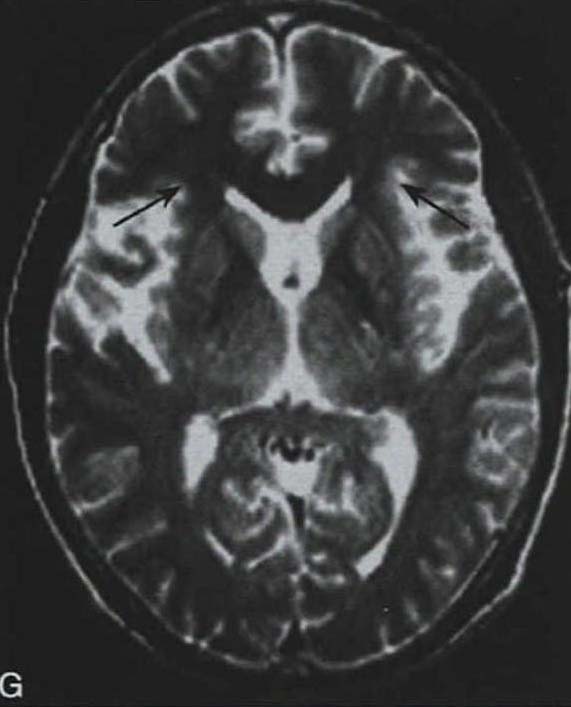 图3-21-4上图,多系统萎缩患者不同病程阶段的脑桥“十字征”分期,从左至右依次为Ⅰ期、Ⅱ期、Ⅲ期、Ⅳ期;下图.壳核“裂隙征”分期(箭头所示):从左至右依次为Ⅰ期、Ⅱ期、Ⅲ期近来一些MRI新技术用于MSA的早期诊断及鉴别诊断(Seppi et al. 2010,Brooks et al. 2009)。基于体素的形态测量法(VBM)用于检测脑干、小脑脚等结构萎缩程度,作为MSA疾病进展的标志。弥散加权像(DWI)显示MSA-P患者壳核弥散度增加,后部受累程度较前部严重,小脑中脚弥散度增高可与进行性核上性麻痹(PSP)鉴别。与健康对照者相比,MSA-C患者脑桥、小脑中脚、小脑白质及壳核ADC值增高。Nilsson等(2007)基于弥散张量成像(diffusion tensor imaging,DTI)和弥散张量纤维示踪图像(diffusion tensor tractology,DTT)研究MSA与PSP及PD进行对比,显示MSA受累部位主要在小脑中脚,PSP主要在小脑上脚,PD患者这两个部位无明显改变(图3-21-5)。磁化传递成像(MTI)显示MSA-P患者存在基底核异常,分析基底核和黑质的磁化传递比率,可将MSA、PSP与PD、健康对照区分开来。经颅超声(TCS)可检测到80%~90%的PD患者中脑黑质高回声信号,至少70%的非典型帕金森综合征(MSA和PSP)患者出现单侧或双侧豆状核高回声信号,而单或双侧豆状核高回声仅见于约25%的PD患者。
图3-21-4上图,多系统萎缩患者不同病程阶段的脑桥“十字征”分期,从左至右依次为Ⅰ期、Ⅱ期、Ⅲ期、Ⅳ期;下图.壳核“裂隙征”分期(箭头所示):从左至右依次为Ⅰ期、Ⅱ期、Ⅲ期近来一些MRI新技术用于MSA的早期诊断及鉴别诊断(Seppi et al. 2010,Brooks et al. 2009)。基于体素的形态测量法(VBM)用于检测脑干、小脑脚等结构萎缩程度,作为MSA疾病进展的标志。弥散加权像(DWI)显示MSA-P患者壳核弥散度增加,后部受累程度较前部严重,小脑中脚弥散度增高可与进行性核上性麻痹(PSP)鉴别。与健康对照者相比,MSA-C患者脑桥、小脑中脚、小脑白质及壳核ADC值增高。Nilsson等(2007)基于弥散张量成像(diffusion tensor imaging,DTI)和弥散张量纤维示踪图像(diffusion tensor tractology,DTT)研究MSA与PSP及PD进行对比,显示MSA受累部位主要在小脑中脚,PSP主要在小脑上脚,PD患者这两个部位无明显改变(图3-21-5)。磁化传递成像(MTI)显示MSA-P患者存在基底核异常,分析基底核和黑质的磁化传递比率,可将MSA、PSP与PD、健康对照区分开来。经颅超声(TCS)可检测到80%~90%的PD患者中脑黑质高回声信号,至少70%的非典型帕金森综合征(MSA和PSP)患者出现单侧或双侧豆状核高回声信号,而单或双侧豆状核高回声仅见于约25%的PD患者。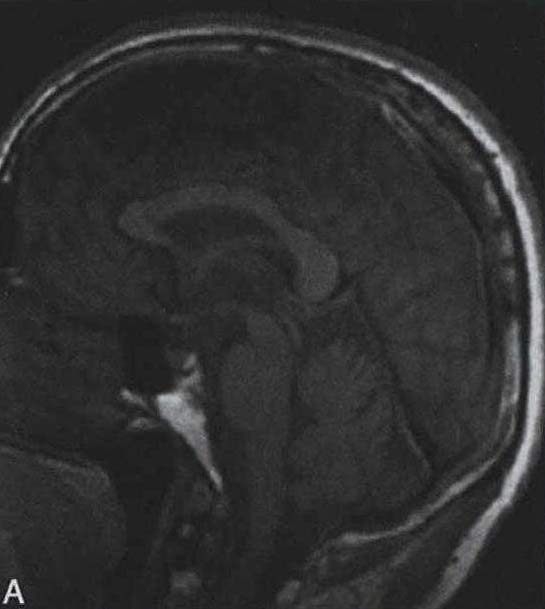
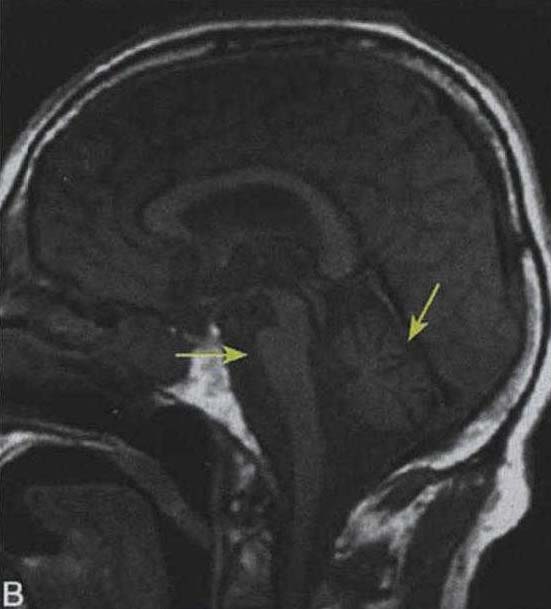
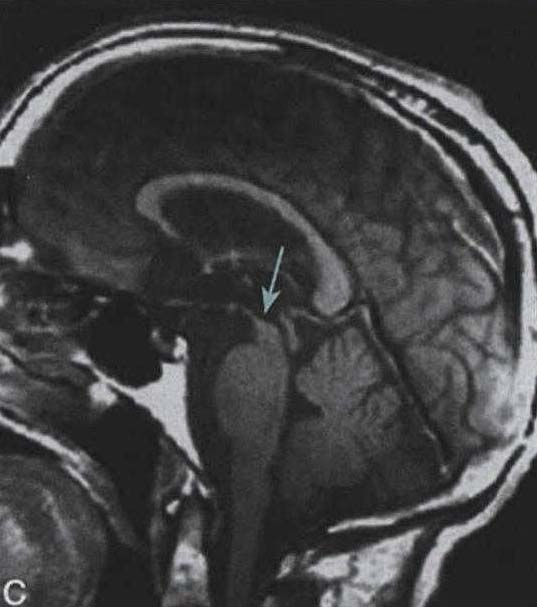

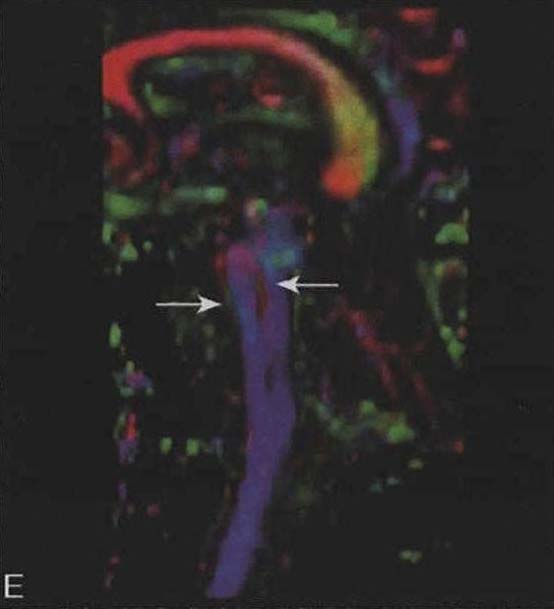



 图3-21-5 特发性帕金森病(IPD)、多系统萎缩(MSA)及进行性核上性麻痹(PSP)矢状位神经影像上图为MRI的T1WI;中图为弥散张量成像(DTI),红色信号显示水平纤维,蓝色信号显示垂直纤维;下图为弥散张量纤维示踪图像(DTT),黄色信号显示小脑中脚纤维,粉色信号显示小脑上脚纤维。左列为IPD患者的影像,未见明显异常;中列为MSA患者的影像,常规MRI显示脑桥小脑萎缩,DTI显示桥横纤维减少,DTT显示小脑中脚明显萎缩;右列为PSP患者的影像,常规MRI显示中脑萎缩呈“鸟嘴征”,DTI未见明显异常,DTT显示小脑上脚明显萎缩功能影像学氟脱氧葡萄糖PET(FDG-PET)显示,MSA患者小脑、脑干、纹状体及额叶皮质局部脑葡萄糖代谢水平下降。PET和SPECT的突触前及突触后多巴胺能成像有助于MSA-P与PD鉴别,MSA患者尾状核受累更严重,D2受体水平下降,而PD患者D2受体水平正常或略升高;绝大多数MSAC患者出现亚临床的黑质纹状体功能障碍。2.肛门括约肌肌电图 MSA患者可出现程度不同的神经源性损害,如平均时限延长、自发电位、纤颤电位、正锐波及波幅增高等。目前认为,这些与骶髓前角细胞中Onuf核弥漫性脱失,导致括约肌的横纹肌失神经支配(Vodusek et al.2001)。肛门括约肌EMG检查有助于MSA诊断,但导致此EMG异常的干扰因素很多,解释异常结果需慎重。王含等(2011)对60例PD患者、68例主要表现PD症状的多系统萎缩(MSA-P)患者及13例PSP患者的肛门括约肌EMG检查显示,肛门括约肌损害在MSA-P较常见和程度较重,在PD较少见和程度较轻,PSP介于两者之间。平均时限延长程度的分布或许可提示骶髓Onuf核在三种疾病中受累程度的差异。3. 自主神经检查 可帮助了解心血管、汗腺分泌、排尿等自主神经功能受损。心脏交感神经节后纤维功能成像对了解心血管系统自主神经功能有一定价值。123I-间碘苄胍(123I-MIBG)心肌显像有助于区分自主神经功能障碍是交感神经节前或节后病变。大多数研究均显示帕金森病组患者心肌摄取123I-MIBG能力降低,多系统萎缩组及对照组无此改变(Orimo et al. 2008)。膀胱功能评价有助于发现早期神经源性膀胱功能障碍,如通过尿动力学试验可检测逼尿肌反射兴奋性升高,尿道括约肌功能减退,疾病后期还可检测到残余尿量增加。膀胱超声检查有助于判断膀胱排空情况,残余尿量>100ml提示膀胱排空障碍,MSA患者膀胱排空障碍常呈进行性加重。4.遗传学检测 MSA目前被定义为散发性神经退行性疾病,但不同患者的表型存在差异,临床上需要与家族史不详、发病年龄晚的遗传性共济失调患者鉴别,有必要筛除常见的脊髓小脑共济失调致病基因动态突变,如SCA1、SCA2、SCA3、SCA6、SCA17等。脆性X染色体震颤/共济失调综合征(Fragile X tremor/ataxia syndrome,FXTAS)表型与MSA有相似之处,可通过遗传学检测FMR1前突变进行鉴别(Garland et al.2004)。
图3-21-5 特发性帕金森病(IPD)、多系统萎缩(MSA)及进行性核上性麻痹(PSP)矢状位神经影像上图为MRI的T1WI;中图为弥散张量成像(DTI),红色信号显示水平纤维,蓝色信号显示垂直纤维;下图为弥散张量纤维示踪图像(DTT),黄色信号显示小脑中脚纤维,粉色信号显示小脑上脚纤维。左列为IPD患者的影像,未见明显异常;中列为MSA患者的影像,常规MRI显示脑桥小脑萎缩,DTI显示桥横纤维减少,DTT显示小脑中脚明显萎缩;右列为PSP患者的影像,常规MRI显示中脑萎缩呈“鸟嘴征”,DTI未见明显异常,DTT显示小脑上脚明显萎缩功能影像学氟脱氧葡萄糖PET(FDG-PET)显示,MSA患者小脑、脑干、纹状体及额叶皮质局部脑葡萄糖代谢水平下降。PET和SPECT的突触前及突触后多巴胺能成像有助于MSA-P与PD鉴别,MSA患者尾状核受累更严重,D2受体水平下降,而PD患者D2受体水平正常或略升高;绝大多数MSAC患者出现亚临床的黑质纹状体功能障碍。2.肛门括约肌肌电图 MSA患者可出现程度不同的神经源性损害,如平均时限延长、自发电位、纤颤电位、正锐波及波幅增高等。目前认为,这些与骶髓前角细胞中Onuf核弥漫性脱失,导致括约肌的横纹肌失神经支配(Vodusek et al.2001)。肛门括约肌EMG检查有助于MSA诊断,但导致此EMG异常的干扰因素很多,解释异常结果需慎重。王含等(2011)对60例PD患者、68例主要表现PD症状的多系统萎缩(MSA-P)患者及13例PSP患者的肛门括约肌EMG检查显示,肛门括约肌损害在MSA-P较常见和程度较重,在PD较少见和程度较轻,PSP介于两者之间。平均时限延长程度的分布或许可提示骶髓Onuf核在三种疾病中受累程度的差异。3. 自主神经检查 可帮助了解心血管、汗腺分泌、排尿等自主神经功能受损。心脏交感神经节后纤维功能成像对了解心血管系统自主神经功能有一定价值。123I-间碘苄胍(123I-MIBG)心肌显像有助于区分自主神经功能障碍是交感神经节前或节后病变。大多数研究均显示帕金森病组患者心肌摄取123I-MIBG能力降低,多系统萎缩组及对照组无此改变(Orimo et al. 2008)。膀胱功能评价有助于发现早期神经源性膀胱功能障碍,如通过尿动力学试验可检测逼尿肌反射兴奋性升高,尿道括约肌功能减退,疾病后期还可检测到残余尿量增加。膀胱超声检查有助于判断膀胱排空情况,残余尿量>100ml提示膀胱排空障碍,MSA患者膀胱排空障碍常呈进行性加重。4.遗传学检测 MSA目前被定义为散发性神经退行性疾病,但不同患者的表型存在差异,临床上需要与家族史不详、发病年龄晚的遗传性共济失调患者鉴别,有必要筛除常见的脊髓小脑共济失调致病基因动态突变,如SCA1、SCA2、SCA3、SCA6、SCA17等。脆性X染色体震颤/共济失调综合征(Fragile X tremor/ataxia syndrome,FXTAS)表型与MSA有相似之处,可通过遗传学检测FMR1前突变进行鉴别(Garland et al.2004)。- +诊断
诊断:MSA的确诊需要病理证实神经胶质胞质包涵体(GCI),伴黑质纹状体及橄榄脑桥小脑通路变性病变,由于临床难以开展CNS活检,因此患者生前难以确诊。这需要医师充分了解该病的特征,临床上细致询问病史及神经系统查体,神经影像学检查,对患者进行长期密切随访,了解疾病的自然病程等颇为重要。由于MSA病因未明,在询问病史时应注意了解可能的危险因素,如长期大量饮酒、毒物接触、CO中毒史及家族史等。1.诊断标准 Gilman等(1998)根据自主神经功能及排尿功能障碍、帕金森综合征、小脑功能障碍及皮质脊髓束损害等四种功能障碍及其特征制定了MSA诊断标准(表3-21-3):基于各功能障碍的组合及其严重程度,将诊断分为可能的、很可能的及确诊的三个级:表3-21-3 Gilman等(1998)诊断标准中MSA功能障碍及其特征功能障碍 特征 标准 自主神经功能和排 1.体位性低血压(收缩压下降20mmHg 体位性低血压(收缩压下降30mmHg或 帕金森综合征 1.运动迟缓(随意动作减慢伴重复动作 运动迟缓加2~4中的一项 小脑功能障碍 1.步态共济失调(步基增宽,步距和方 步态共济失调加2~4中的一项 皮质脊髓束损害 伸性足跖反射伴腱反射亢进 不需要皮质脊髓束损害特征 (1) 可能的(possible) MSA:符合1项功能障碍的诊断标准和另外的不同功能障碍的2个特征,当诊断标准为帕金森综合征时,对多巴胺反应差可作为一个特征,此时仅需要有另一特征即可。(2) 很可能的(probable) MSA:为自主神经功能及排尿功能障碍诊断标准,加上多巴胺反应差的帕金森综合征或小脑功能障碍的特征。(3) 确诊的(definite)MSA:为病理证实的神经胶质胞质包涵体(GCI),伴黑质纹状体和橄榄脑桥小脑通路变性改变。排除标准包括发病年龄<30岁,有类似的家族史,存在系统性疾病或其他明确原因导致的MSA特征以及与药物无关的幻觉;神经系统检查显示符合痴呆的DSM标准,垂直快速扫视明显减慢或垂直性核上性凝视麻痹,局部皮质病变证据如失语、异肢症、顶叶功能障碍等。2008年Gilman等根据近10年对MSA的临床研究对诊断标准进行了修订,限定MSA为散发性、进展性、成人起病(>30岁)的神经变性病。很可能的MSA为符合标准的自主神经功能障碍伴左旋多巴反应差的帕金森综合征或小脑功能障碍。修订版诊断标准主要增加了“可能的诊断”,定义为帕金森综合征或小脑功能障碍合并至少一项自主神经功能障碍和至少一项其他特征(表3-21-4),同时增加了支持/不支持特征(表3-21-5)以便进行早期诊断及鉴别诊断。表3-21-4 可能的MSA的其他特征可能的MSA-P或MSA-C 可能的MSA-P 可能的MSA-C 1. Babinski征伴腱反射亢进 1.进展较快的帕金森综合征 1.帕金森综合征(运动迟缓和强 表3-21-5 MSA支持/不支持的特征支持的特征 不支持的特征 1.口面部的肌张力障碍 1.具有代表性的搓丸样震颤 2.评估量表 欧洲多系统萎缩研究组(EMSASG)于2004年建立了统一的多系统萎缩评估量表(unified multiple system atrophy rating scale,UMSARS),以Hoehn-Yahr帕金森病分级,Schwab and England量表(SES),UPDRS,ICARS和复合自主神经系统量表等作为模板,主要包括病史回顾、运动检查、自主神经功能检查和整体失能程度评分,每一项目定义了0分(正常)到4分(严重异常)的特征。通过在欧洲多个神经中心使用,显示该量表能较好地反映疾病严重程度的变化(Wenning et al.2004)。顾卫红等(2007,2008)在获得量表作者授权后将UMSARS翻译为中文,对一组中国MSA患者进行了UMSARS与病程相关性分析。- +鉴别诊断
鉴别诊断:不同的MSA患者在发病初期可分别表现为帕金森病、橄榄脑桥小脑萎缩或纯自主神经衰竭(pure autonomic failure,PAF),需要进行鉴别。在病史方面,快速动眼期睡眠行为障碍在MSA较显著,且大多出现于发病之前。随访对鉴别诊断也很重要。研究显示,最初诊断为帕金森病的患者中9%~20%最终进展为MSA;29%~33%散发的晚发性共济失调进展为MSA(Köllensperger et al.2010)。MSA需要与以下疾病鉴别:(1) 帕金森病:MSA-P发病初期表型类似PD,但左旋多巴疗效不明显,且自主神经功能障碍较明显,随病程进展两者表型的差异逐渐显现,因此需临床随访观察。鉴别要点是:①MSA-P患者对左旋多巴反应差;②MSA进展较快,姿势反射异常出现早;③自主神经功能异常出现早,且较显著,包括体位性低血压、出汗异常、尿便障碍等;④神经影像学可显示MSA患者脑桥、小脑和壳核萎缩;⑤经颅超声可显示PD患者黑质高回声区,而MSA-P患者黑质区异常回声不明显,可出现单侧或双侧豆状核高回声信号。(2) 橄榄脑桥小脑萎缩(OPCA):最初对一例共济失调患者的病理特征描述为下橄榄体、脑桥基底部及小脑中脚变性,此后不同研究者对共济失调疾病进行了大量临床及病理研究,根据表型进行分类,人们逐渐将散发性与遗传性小脑功能异常(尤其常染色体显性遗传)疾病归于OPCA。目前,广义的OPCA包括散发的晚发性共济失调、MSA-C和SCA;狭义的OPCA仅指散发的晚发性共济失调,与MSAC的主要区别是,MSA-C除了共济失调,还有明显的自主神经功能异常(Berciano et al.2006)。(3) 纯自主神经衰竭(PAF):Shy-Drager综合征指自主神经功能障碍伴锥体外系或小脑、脑干损害,属于MSA的一种表型。Shy-Drager综合征曾被描述为PAF,但有人认为PAF仅是Shy-Drager综合征的一个病程阶段。近年研究显示两者存在一定的差异,MSA患者早期出现排尿异常,继而出现泌汗异常及体位性低血压,以及呼吸障碍,病程持续进展;PAF患者早期出现头晕和泌汗异常,继而出现便秘和晕厥,排尿障碍出现晚,无明显呼吸障碍,病程进展较慢(Donadio et al.2010)。(4) 脆性X染色体震颤和(或)共济失调综合征(FXTAS):表现进行性小脑共济失调、震颤、智力衰退、帕金森综合征及自主神经异常,与MSA有一定的相似之处,但FXTAS表现更明显,可通过基因检测FMR1基因前突变进行鉴别。(5) 慢性乙醇中毒的共济失调:患者有长期大量饮酒史,表现为步态和下肢共济失调,上肢、语言和眼动症状不明显,自主神经障碍不明显,戒酒后病程进展较MSA缓慢。(6) 副肿瘤综合征:亚急性发病,伴共济失调,患者一般在短时间内体重明显下降,肿瘤相关检测可能有助于鉴别。- +治疗
治疗:多系统萎缩的早期诊断与干预很重要,目前MSA治疗包括对症治疗及神经保护治疗(Wenning et al. 2009, Köllensperger et al. 2010)。1. 对症治疗(1) 体位性低血压:①如果卧位血压偏低或正常,可试用盐酸米多君,每天3次,初始剂量2.5mg,监测卧位、立位血压,以卧位血压<140/90mmHg为限,调整药量(需参考患者发病前血压水平);部分患者服用盐酸米多君后出现尿频,若不能耐受应及时停药,如果卧位血压高于正常,不建议服用盐酸米多君;体位性低血压亦可试用溴吡斯的明,国外文献报道可缩小卧、立位血压变化(Singer et al. 2003);②中药如生脉饮或生脉胶囊有升压作用,中药方剂需因人而异,一般可能引起轻度水钠潴留,部分患者可出现下肢水肿;③注意部分MSA患者存在中枢性低钠,感觉乏力,尽管活动量减少但食欲较好,需增加盐的摄入;其他应注意晨起饮盐水,少食多餐以避免餐后低血压反应,穿弹力袜,卧位坐起或站立时尽可能动作慢些,睡眠时后背倾斜垫高30°,尽量避免炎热环境,血钠明显下降患者可服用氯化钠胶囊;须谨慎用药,某些药物有降血压作用,应避免服用,既往有高血压病患者注意降压药用法和用量;由于左旋多巴可加重体位性低血压,MSA帕金森型患者服用这类药物时要注意用量,权衡利弊。(2) 排尿障碍:MSA患者可见尿频、尿失禁、尿不净、夜尿次数增多。尿频患者可试用艾灸,穴位可选择关元和命门,每侧灸20分钟。尿不净患者必要时可行间断导尿,及时冲洗避免感染。尿失禁可用曲司氯铵(trospium chloride) 20mg,每天2次,或15mg口服,每天3次;也可用奥昔布宁2.5~5mg,每天2~3次,需注意该药中枢性不良作用,或托特罗定2mg口服,每天2次。排尿不净,残余尿>100ml是留置尿管的适应证,晚期患者可行耻骨上膀胱造瘘。抗利尿激素类似物去氨加压素可减少夜尿并改善清晨体位性低血压(Alam et al.1995)。(3) 排便无力:可采用一些中药缓泻剂,按摩腹部,适当增加运动,做提肛运动训练括约肌。(4) 睡眠障碍:REM睡眠行为障碍可试用氯硝西泮0.5mg,睡前服,需注意患者呼吸情况,如已出现睡眠呼吸暂停,应慎用氯硝西泮及其他安眠药,及时到呼吸内科就诊。(5) 僵直和动作迟缓:MSA-P患者虽服用多巴丝肼等药物效果不明显,但国外仍推荐增加多巴丝肼(美多芭)剂量,仍有可能有一定的疗效。需要注意多巴丝肼可能加重体位性低血压,需因人而异调整剂量。(6) 行走不稳:MSA患者姿势反射异常与脑内胆碱能神经递质缺乏有关(Gilman et al. 2010),可尝试拟胆碱药治疗;在改善体位性低血压,保证安全的前提下进行平衡康复训练。2.针对MSA的神经保护治疗临床药物试验(1) 重组人类生长激素(recombinant human growth hormone,r-HGH):基于UPDRS和UMSARS评分显示,r-HGH可延缓MSA患者运动功能衰退速度,但结果未达到统计学显著标准(Holmberg et al.2007)。(2) 米诺环素(minocycline):是一种抗生素,前期基于MSA小鼠模型及其他神经退行性疾病动物模型研究显示有神经保护作用(Stefanova et al.2007),但EMSA-SG开展的Ⅱ期临床试验显示无效(Dodel et al. 2010)。(3) 利鲁唑(riluzole):具有拮抗兴奋性氨基酸谷氨酸的作用,NNIPPS研究(Neuroprotection and Natural History in Parkinson Plus Syndromes)显示对帕金森综合征包括MSA无效(Bensimon et al.2009)。(4) 雷沙吉兰(rasagiline):新型选择性单胺氧化酶B抑制药,基础及临床试验显示有神经保护作用。雷沙吉兰对MSA转基因小鼠模型有神经保护作用,包括运动功能改善,减轻黑质致密部、纹状体、小脑皮质、脑桥核和下橄榄核神经元缺失(Stefanova et al.2008)。(5) 辅酶Q10:作为线粒体呼吸链的组成成分,安全性和对神经退行性疾病的潜在治疗作用已被一些基础研究和临床试验初步证实,包括帕金森病、亨廷顿舞蹈病、阿尔茨海默痴呆、弗里德里希共济失调、运动神经元病等(Spindler et al. 2009)。- +预后
与帕金森病及散发的晚发型共济失调相比,MSA病程进展快,平均病程为8~9年,患者早期出现自主神经功能障碍预后不良。随病程进展,MSA患者大多出现腰部酸痛无法直起,下肢无力,严重姿势反射异常,经常摔倒,导致卧床,晚期可因自主神经功能衰竭在睡眠中出现呼吸骤停死亡。Shimohata等(2008)对45例诊断很可能的日本MSA患者随访5年,10例死亡,其中7例为不明原因猝死,大多在夜间发生,睡眠呼吸暂停可能是主要原因。Watanabe等(2002)总结230例日本MSA患者病程及预后,从发病至需辅助行走、坐轮椅、卧床和死亡的中位数年限分别为3年、5年、8年和9年。相关检查
- 知识库去甲-3-甲氧肾上腺素
- 知识库尿儿茶酚胺
- +诊断